Chapter 23 Primary Retroperitoneal Tumors
Introduction
Primary retroperitoneal tumors are an exceedingly rare clinical problem. Masses in the retroperitoneum can be categorized as one of three entities: lymphomas, extragonadal germ cell tumors, and sarcomas. Although gastrointestinal stromal tumors (GISTs) arise in the intraperitoneal compartment, they can also mimic these retroperitoneal masses because of their large size. This chapter deals with primary retroperitoneal sarcomas (RPSs), which form about one third of all retroperitoneal tumors. Lymphomas, extragonadal germ cell tumors, and GISTs are discussed in Chapters 16, 20, and 29. RPSs generally lack specific clinical symptoms beyond the effect manifested from their mass or impact on adjacent organs. As a result, they progress to a very large size before the patient seeks medical care. Their successful management requires the collaborative efforts of the radiologist, pathologist, radiation oncologist, medical oncologist, surgical oncologist, and other specialists. Imaging plays a key role in the detection, planning of therapy, and follow-up of these patients.
Epidemiology and Risk Factors
Soft tissue sarcomas (STSs) form 1% of all newly diagnosed malignancies in the United States and 15% of these arise in the retroperitoneum.1 STSs in children account for up to 15% of all pediatric malignancies. The mean annual incidence of RPS is 2.7 per million persons and no significant change has been observed in a recent review encompassing 29 years of data.2
Exposure to ionizing radiation increases the incidence of STSs, with a median interval of 10 years (range 2-50 yr) after exposure. These occur in the irradiated field commonly in patients who receive radiation therapy for breast, cervical cancer, lymphoma, and testicular tumors or for benign conditions.3–5 Patients treated with prior radiation exposure can develop either STSs or bone sarcomas.
Several genetic syndromes are associated with the development of STS.6 Neurofibromatosis type 1, or von Recklinghausen’s disease, is an autosomal dominant condition that is due to a mutation in the NF1 gene. These patients have a high incidence of neurofibromas as well as approximately a 10% increased risk of developing malignant peripheral nerve sheath tumors during their lifetime. Gardner’s syndrome, another autosomal dominant disease, is caused by mutation of the APC gene and associated with multiple colonic polyps, colon cancer, and desmoid tumors. STS has also been reported in patients with the Li-Fraumeni syndrome, caused by a germline mutation in the p53 tumor-suppressor gene. Children with hereditary retinoblastoma due to a germline mutation in the RB1 tumor-suppressor gene face a higher risk of STS and osteosarcoma. The risk is further increased because these patients receive radiotherapy for the initial treatment of retinoblastoma.
Anatomy and Pathology
Anatomy
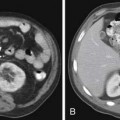
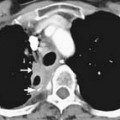
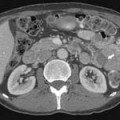
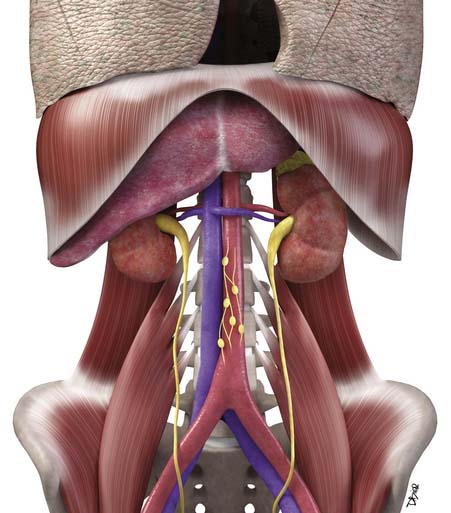
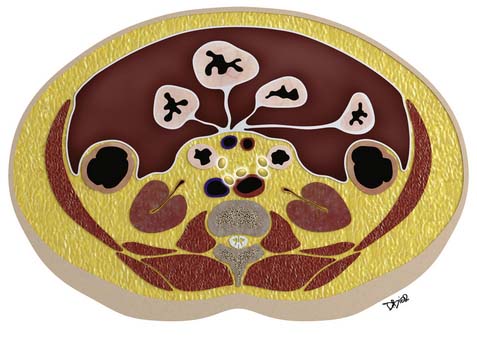
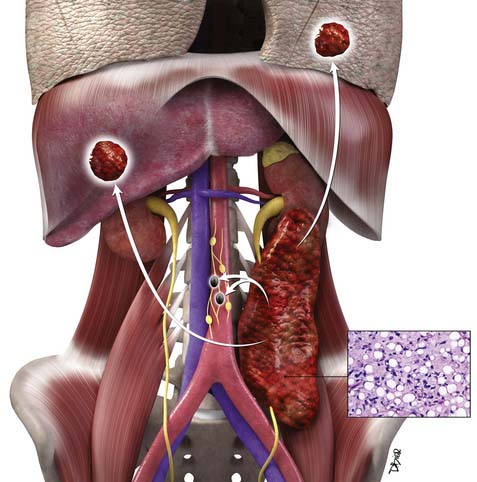
Macroscopically, parts of the genitourinary tract, gastrointestinal (GI) tract, aorta and its branches, inferior vena cava and its tributaries, and lymphatic and nervous systems form important components of the retroperitoneum (Figure 23-1). The pancreas, ascending colon, descending colon, and duodenum are located anteriorly within the anterior pararenal space (APS), and the aorta, inferior vena cava, and lymph nodes are in the midline. Laterally, the kidneys and adrenals are surrounded by renal fascia within the perirenal space (PS). The psoas, quadratus lumborum, paraverterbral muscles, and bony skeleton form the posterior boundary of the retroperitoneum. Retroperitoneal fat, vessels, lymphatics, and nerves continue from the retroperitoneum into the small bowel mesentery, providing anatomic continuity between these compartments (Figure 23-2). The displacement or contiguous involvement of these major organs, and in particular of the major vascular branches, is of critical importance when planning surgical resection.
Key Points Anatomy
• Duodenum, ascending and descending colon, and pancreas lie anteriorly in the APS.
• Kidneys and adrenals surrounded by renal fascia laterally lie within the perirenal space.
• Muscles and bones form the posterior boundary of the retroperitoneum.
• Arterial encasement, venous involvement, and the displacement or invasion of adjacent organs are important considerations in surgical planning.
Pathology
Microscopically, primary sarcomas can arise from fat, smooth or skeletal muscle, fibrous connective tissue, peripheral nerve cells, vascular tissue, or other mesenchymal cells (Table 23-1). STSs are classified according to the adult cell type that the tumor cells most closely resemble. However, this does not mean that the tumor arose from that cell type. The use of immune markers provides important additional information in the classification of STS. Liposarcomas are the most common type of tumor in adults, followed by leiomyosarcoma.7 In the older reports, the category of “malignant fibrous histiocytoma (MFH)” was common, whereas in current series, it is distinctly uncommon and classified instead as “undifferentiated pleomorphic sarcoma.”8 Ten percent to 15% of all sarcomas occur in children. In reviews from single institutions, the most common retroperitoneal sarcoma in the pediatric age is rhabdomyosarcoma, followed by fibrosarcoma and liposarcoma.9,10
Table 23-1 World Health Organization Classification of Soft Tissue Sarcomas
| Adipocytic Tumors |
| Atypical lipomatous tumor (ALT)/well-differentiated liposarcoma (WDLPS) |
| Dedifferentiated liposarcoma (DDLPS) |
| Myxoid/round cell liposarcoma |
| Pleomorphic liposarcoma |
| Smooth Muscle Tumors |
| Leiomyosarcoma |
| Skeletal Muscle Tumors |
| Rhabdomyosarcoma (embryonal, alveolar, and pleomorphic) |
| Fibroblastic/Myofibroblastic Tumors |
| Fibrosarcoma |
| Low-grade myxofibrosarcoma |
| Low-grade fibromyxoid sarcoma |
| Sclerosing epithelioid fibrosarcoma |
| So-called Fibrohistiocytic Tumors |
| Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma (MFH) (including pleomorphic, giant cell, myxoid high-grade myxofibrosarcoma, and inflammatory forms) |
| Tumors of Peripheral Nerves |
| Malignant peripheral nerve sheath tumor |
| Vascular Tumors |
| Epithelioid hemangioendothelioma |
| Deep angiosarcoma |
| Chondro-osseous Tumors |
| Extraskeletal chondrosarcoma or osteosarcoma |
| Tumors of Uncertain Differentiation |
| Synovial sarcoma |
| Epithelioid sarcoma |
| Alveolar soft part sarcoma |
| Clear cell sarcoma of soft tissue |
| Extraskeletal myxoid chondrosarcoma |
| PNET/extraskeletal Ewing’s tumor |
| Desmoplastic round cell tumor |
| Extrarenal rhabdoid tumor |
| Undifferentiated sarcoma |
MFH, malignant fibrous histiocytoma; PNET, primitive neuroectodermal tumor.
From Fletcher C, Unni KK, Mertens F. Pathology and Genetics of Soft Tissue and Bone: World Health Organization Classification of Tumors. Lyon, France: IARC Press; 2002.
Liposarcomas are tumors composed of fat cells. The most common form that arises in the retroperitoneum is well-differentiated liposarcoma (WDLPS). Morphologically, it is similar to a typical lipoma; histologically, the presence of lipoblasts allows its recognition. WDLPSs show slow but progressive growth over many years without development of any metastasis. In approximately 25% of the cases, there is transformation into a higher grade of tumor. When dedifferentiation occurs, there is loss of mature fat, and the tumor grows faster and has the capacity to metastasize. These areas of dedifferentiated liposarcomas (DDLPSs) always occur in preexisting WDLPS and show an abrupt transition to a nonfat component. Sometimes, these areas that show dedifferentiation may display features of leiomyosaroma or other sarcoma on histology and by immune markers. Dedifferentiation is characterized by more aggressive local growth, a greater risk of recurrence after resection, and development of distant metastases in 15% to 30%. The myxoid/round cell and pleomorphic subtypes of liposarcomas are uncommon in the retroperitoneum.11
The common STSs in children are age-dependent. Up to age 14, rhabdomyosarcoma is the most common tumor type, whereas nonrhabdomyosarcomas are common in adolescents and young adults.10,12 In a single institutional report covering 30 years that specifically looked at retroperitoneal site of tumor origin, rhabdomyosarcoma, followed by fibrosarcoma, were reported as common types of RPSs in the pediatric age group.9 This pattern is distinctly different from common tumor histology in adults. The common subtype of rhabdomyosarcoma is embryonal arising in the genitourinary system. These tumors are large, infiltrative, and liable to involve adjacent organs at the time of initial presentation. Thus, positive margins may be present after gross resection of tumor. Microscopically, these tumor cells do not arise from skeletal myoblasts but rather resemble them.11
Clinical Presentation
RPSs lack specific symptoms or laboratory findings. They usually progress to a very large size before prompting the astute clinician to consider this diagnosis. A tip off can be a patient who is discovered to have a painless abdominal mass. Other presenting symptoms may be abdominal distention, back pain or pain referred to the hip, urine retention, hematuria, early satiety, GI obstruction, or weight loss, occurring singly or in combination. Neurologic deficits may be a presenting symptom when there is spinal cord, nerve root, or sciatic plexus involvement. Venous compromise can result in lower extremity edema. Although there is a broad age range, the sixth decade is a common time for presentation in adults. There is slight male predominance. Imaging, typically abdominal and pelvic computed tomography (CT), leads to discovery of the tumor. Eleven percent of patients with RPS at presentation have metastases to liver or lungs.14,15 Two thirds of the tumors are high grade at the time of diagnosis.14,15 Therefore, at their initial presentation, RPSs are at a higher stage and, consequently, their prognosis is worse than for extremity sarcomas.
Staging Evaluation
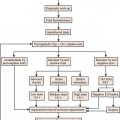
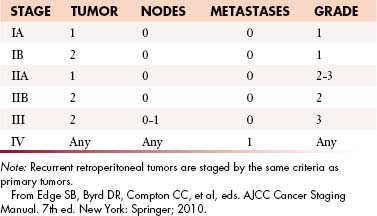
The current American Joint Committee on Cancer (AJCC) staging system for STS is based on a combination of anatomic as well as pathologic data: (1) tumor size, (2) depth, (3) histologic type, and (4) grade16 (Table 23-2). It has been derived from experience based on sarcomas of the extremities and applied to all sites including the retroperitoneum. The tumor-node-metastasis (TNM) characteristics of the tumor are provided by imaging and may be modified after surgery. The histologic subtype and grade of the primary tumor are determined after biopsy or surgical excision to yield the stage of tumor by AJCC criteria.
Pathologic Criteria: Grading
1. Histology-specific differentiation: high, intermediate, or low grade. Also certain histologic types (synovial sarcoma, undifferentiated sarcoma, and sarcomas of unknown type) are always assigned a high score,
This combination of T, N, M, and G for AJCC staging is depicted in Figure 23-3.
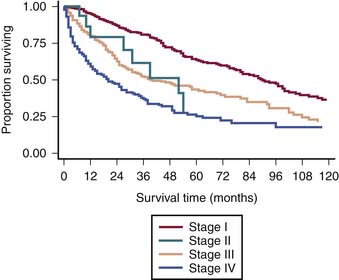
The survival graph shows better survival at lower stages (Figure 23-4). However, the absence of a major difference in survival between stage II (N0 disease) and stage III (N1 disease) points to a limitation in the current AJCC staging for RPSs (Figure 23-5).
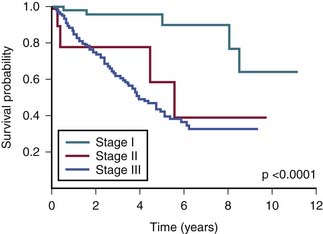
Limitations of American Joint Committee on Cancer Classification
Site of Origin
The AJCC classification system does not differentiate retroperitoneal origin from extremity or other sites of origin. However, this does affect the stage at presentation and subsequent outcome.19 Therefore, when outcome data are being evaluated, it is important to consider RPSs as distinct from extremity, head and neck, gynecologic, and other sites of origin.
Size of Primary Tumor
The limitation of using an arbitrary 5 cm as the cutoff has been shown for retroperitoneal tumors that tend to be very often large at presentation. It is recognized that tumor size represents a continuous variable: the greater the size at initial presentation, the worse the outcome.19
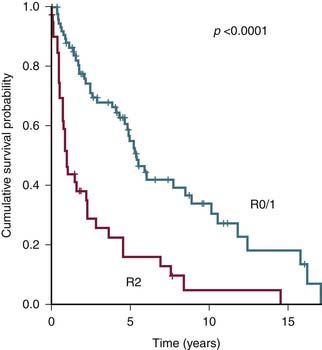
Margin Status of Resected Tumor
The status of resected margin after surgery is an important prognostic factor but not a part of the AJCC staging20–22 (Figure 23-6).
Other Staging Systems
The most important common variables in the AJCC system for RPSs are the presence of distant metastasis M1 and the tumor grade G1-3. Therefore, alternate staging systems based only on tumor grade or with additional criteria of multifocality and the degree of residual tumor left after surgery (R0, R1, or R2) have been proposed and are as good as or even better than AJCC.17,18
Today, we are poised at the brink of important change with the availability of high-throughput technology of genomic, proteomic, and tissue microarray analysis in sarcoma. As the data about relevant genes and molecular markers are integrated with disease progression, tumor response, and survival, future modifications can be made to AJCC staging.