ANATOMY AND PHYSIOLOGY OF THE PULMONARY CIRCULATION
The pulmonary circulation consists of two parallel networks: the pulmonary arterial circulation and the bronchial arterial circulation. Pulmonary arteries course along the lobar, segmental, and subsegmental airways to the level of the terminal bronchioles. Small pulmonary arteries from the subsegmental level to the terminal bronchioles possess a thick muscular media, and range from 50 to 1,000 μm in size. These small pulmonary arteries progressively lose much of their muscle within the arteriolar media as well as their external elastic membrane. By the level of the respiratory bronchioles and alveolar ducts they are termed pulmonary arterioles, and range in size from 10 to 150 μm. These vessels ramify further within the alveolar walls to form a rich capillary network. Capillary blood collects in venules, which coalesce progressively to form veins, which course within the interlobular septa, eventually to empty into the left atrium.
The bronchial circulation, accounting for about 1% of the systemic cardiac output, originates from the thoracic aorta or intercostal arteries. Bronchial arteries, averaging two per lung, course within the pulmonary hila along the mainstem bronchi to the level of the terminal bronchiole and form a plexus that extends from the adventitia through to the submucosa of the associated airway. Bronchial arteries freely form anastomoses with pulmonary arteries, primarily at the capillary and postcapillary levels.
Unlike the tracheobronchial system, in which the major component to airflow resistance is located within the large airways, the major site of resistance to pulmonary arterial blood flow is located at the small muscular pulmonary arterial and arteriolar level. Caliber changes in the vessels at this level regulate pulmonary arterial pressure and are critical for optimizing ventilation and perfusion matching.
The pulmonary circulation is a low pressure system—the mean arterial pressure is approximately one sixth that of the systemic circulation. This low pressure is maintained at a relatively consistent level even with large increases in pulmonary blood flow such as may occur with exercise. This is possible because when the body is at rest, numerous pulmonary capillaries normally are not perfused; these capillaries are “recruited” when increased pulmonary blood flow must be accommodated.
PATHOGENESIS OF PULMONARY HYPERTENSION
Pulmonary hypertension (PH) is defined as a resting mean pulmonary arterial pressure (mPAP) ≥25 mm Hg at rest or 30 mm Hg with exercise. The subgroup of PH referred to as pulmonary arterial hypertension (PAH) adds the criterion that the pulmonary capillary wedge pressure is ≤15 mm Hg. Pulmonary venous hypertension is present when pulmonary venous pressure, usually approximated by measurement of the pulmonary capillary wedge pressure, is equal to or exceeds 18 mm Hg. Several mechanisms may produce a decrease in the total number of small pulmonary arteries, thereby increasing pulmonary vascular resistance and producing elevated pulmonary arterial pressure. These mechanisms include intraluminal arterial occlusion, muscular contraction of small pulmonary arteries, vascular remodeling with wall thickening, or conditions that produce pulmonary venous hypertension. Several of these mechanisms may be operative simultaneously in a patient with pulmonary hypertension.
The pulmonary vascular endothelium responds to changes in oxygen tension, transmural pressure, and pulmonary blood flow and participates actively in the regulation of pulmonary arterial pressure through the elaboration of various vasoactive substances, such as prostacyclin, nitrous oxide, and endothelin. The agents have a direct effect on pulmonary vascular smooth muscle tone (promoting relaxation and vasodilation) and also may directly affect platelet function. Abnormalities in endothelial cell function or injuries to these cells may be the fundamental derangement that ultimately produces the structural vascular changes observed in patients with pulmonary hypertension.
Various histopathological abnormalities may be observed in patients with pulmonary hypertension, varying somewhat depending on the cause of hypertension. In general, regardless of the specific cause of the pulmonary hypertension, the pulmonary arteries become dilated, occasionally to the point of being considered aneurysmal. Pulmonary arterial atherosclerosis, although occasionally present to a mild degree in the larger pulmonary arteries of normal adults, often is extensive in patients with pulmonary hypertension and commonly involves small arteries. Pulmonary hypertension-related pulmonary arterial atherosclerosis pathologically appears similar to atherosclerosis in systemic arteries, although complicating features, such as necrosis, ulceration, and calcification, are relatively uncommon. Thickening of the muscular media of small pulmonary arteries is a common feature in many causes of pulmonary hypertension and usually results from a combination of muscular hyperplasia and hypertrophy. Often, extension of muscular tissue into arterioles that normally contain no muscle, or “arterialization,” may be observed in patients with pulmonary hypertension.
The term pulmonary plexogenic arteriopathy refers to a constellation of histopathological vascular changes that often is encountered in patients with PAH, including patients hepatic disease, connective tissue disorders, congenital cardiovascular disease, and some medications previously prescribed for weight loss. Less commonly, plexogenic arteriopathy may be encountered in patients with other causes of pulmonary hypertension. Histopathological features present in pulmonary plexogenic arteriopathy include a combination of fibrinoid necrosis, dilation lesions, plexiform lesions, intimal fibrosis, and vasculitis. Plexiform lesions affect small muscular arteries ranging in size from 100 to 200 μm, usually near vascular branch points, and consist of a focally dilated muscular vessel with a disrupted internal elastic membrane that contains very narrow vascular channels interspersed with fibroblasts and connective tissue. Plexiform lesions are characteristic of prolonged severe pulmonary hypertension.
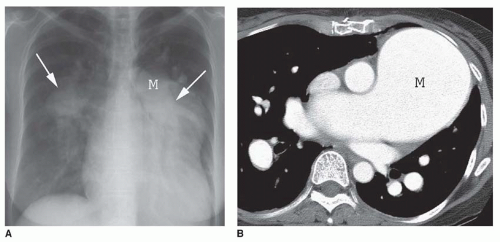
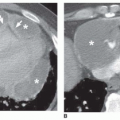
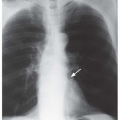
FIG. 28.1. Pulmonary hypertension: pulmonary arterial enlargement. A: Frontal chest radiograph shows massive enlargement of the main pulmonary artery (M) and bilateral interlobar pulmonary arteries (arrows). B: CT shows massive enlargement of the main pulmonary artery (M).
GENERAL IMAGING MANIFESTATIONS
The characteristic finding of pulmonary hypertension on chest radiography, CT, or MRI is dilation of the central pulmonary arteries with rapid tapering of the pulmonary vessels as they course peripherally (Fig. 28-1). This pattern is present regardless of the etiology of the pulmonary hypertension.
Chest radiography may reveal enlargement of the main pulmonary artery segment and dilation of the right and left main and interlobar pulmonary arteries in patients with pulmonary hypertension of any etiology. It has been suggested that pulmonary hypertension may be diagnosed on chest radiography if the transverse diameter of the right interlobar pulmonary artery, measured from the lateral aspect of the vessel to its medial portion adjacent to the bronchus intermedius, exceeds 15 mm in women and 16 mm in men. Similarly, an enlarged left pulmonary artery also may indicate the presence of pulmonary hypertension. The left pulmonary artery is best measured on the lateral radiograph from the orifice of the left upper lobe bronchus to the posterior aspect of the vessel; when this measurement exceeds 18 mm, pulmonary hypertension probably is present.
The main pulmonary arterial segment cannot be measured on chest radiography, but it is measured easily on CT or MRI. The upper size limit for a normal main pulmonary arterial segment on axial CT or MR images is 29 mm. When the main pulmonary artery segment exceeds this size (see Fig. 28-1B), pulmonary hypertension is usually, but not invariably, present. Furthermore, pulmonary hypertension may be present in patients with a main pulmonary arterial segment that is normal in size. Rather than measuring the main pulmonary artery, one may compare the size of the main pulmonary artery with the ascending aorta near the base of the heart. If the main pulmonary artery is visibly larger than the aorta, elevated pulmonary pressures usually are present.
When pulmonary hypertension is prolonged and severe, calcification of the pulmonary arteries, usually affecting the main, right, or left pulmonary arteries, and, less commonly, the lobar pulmonary arteries, may be present. This finding usually, but not invariably, is associated with irreversible vascular disease.
Although chest radiography and CT scanning often may suggest the presence of pulmonary hypertension, echocardiography is the examination most commonly used for noninvasive assessment of possible pulmonary hypertension. Echocardiography, using continuous wave or pulsed Doppler, provides noninvasive estimation of pulmonary arterial pressures and also allows detailed morphologic evaluation of the right ventricle. Echocardiography is also very useful for the assessment of left ventricular morphology and function as well as valvular function. Echocardiography also is used for the assessment of the hemodynamic changes in the pulmonary arterial circulation in response to a variety of challenges, such as exercise or pharmacologic agents; this technique is termed stress echocardiography. Echocardiography does have some limitations, particularly difficulty obtaining appropriate acoustic windows in patients with emphysema, as well as operator dependence. Also, the complex anatomical shape of the right ventricle can make the echocardiographic assessment of right ventricular dimensions challenging.
MRI can provide functional information equivalent to that available from echocardiography (including stress echocardiography), such as direction and velocity of blood flow, in addition to specific anatomic information. MR techniques are well suited for the evaluation of patients with pulmonary hypertension because they allow both a detailed anatomic and an extensive functional examination of the cardiovascular system in general and right ventricular performance in particular. MR is assuming a greater role in the initial assessment and serial evaluation of patients with pulmonary hypertension since the primary clinical outcome in these patients, progressive right heart failure, can be readily evaluated with MRI. The complex shape of the right ventricle does not adversely affect the measurement of chamber dimensions with MR, and MR provides superior spatial resolution in comparison with echocardiography. MR provides reliable functional data with excellent interstudy reproducibility for functional measurements and anatomic dimensions.
CLASSIFICATION OF PULMONARY HYPERTENSION
Numerous classification schemes have been developed to categorize the causes of and associations with pulmonary hypertension. One method has been to examine the disease from a physiologic perspective, using the relations among pressure, pulmonary vascular resistance, and pulmonary flow. In this type of classification, diseases that cause increased resistance, increased flow, or increased pulmonary vascular pressure are grouped separately. In another scheme, pulmonary hypertension was classified into two broad categories: primary pulmonary hypertension and secondary pulmonary hypertension, based on the presence or absence of identifiable risk factors or causes for pulmonary hypertension. Other investigators have classified pulmonary hypertension into precapillary and postcapillary etiologies (Table 28-1). In 1998, during the Second World Symposium on Pulmonary Hypertension held in Evian, France, a new clinical classification system for pulmonary hypertension emerged. The impetus for the new classification system for pulmonary hypertension was to provide a scheme that groups diseases and conditions associated with pulmonary hypertension according to similarities in the pathophysiologic mechanisms, clinical presentation, and treatment options. Such a classification system has obvious advantages for standardizing diagnostic approaches, conducting clinical trials, determining the effectiveness of therapy, and assessing prognosis in patients with pulmonary hypertension. Subsequently, the Third World Symposium on Pulmonary Arterial Hypertension held in Venice, Italy, in 2003, allowed assessment of the impact of the Evian classification and provided the opportunity for further refinement. Subsequently, the 4th World Symposium on Pulmonary Hypertension, held in Dana Point, California, in 2008, confirmed the utility of the Evian-Venice classification systems, but revisions to the classification system for pulmonary hypertension were proposed to reflect additional information published over the previous 5 years. The Dana Point Classification system for pulmonary hypertension is presented in (Table 28-2), and it recognizes five major categories of pulmonary hypertension. The Dana Point pulmonary hypertension classification system is used in this chapter since it reflects the most comprehensive and advanced approach to the diagnosis, assessment, and treatment of pulmonary hypertension and is in common use by experts in the field, not to mention the physicians referring patients to radiologists for the imaging assessment of pulmonary hypertension. Although seemingly complex and distinct compared to pulmonary hypertension classification systems previously used in the radiology literature, the five major disease classifications in the 2008 Dana Point classification broadly group the causes of pulmonary hypertension in a fashion that is relevant to imaging and has the added benefit of providing subclassifications that serve as a reminder that pulmonary hypertension can be associated with a number of conditions affecting various organ systems.
TABLE 28.1 Precapillary and Postcapillary Classification of Pulmonary Hypertension
Precapillary Etiologies
Primary Pulmonary hypertension
Pulmonary hypertension associated with
Hepatic disease
HIV infection
Drugs and toxins
Congenital cardiovascular disease
Chronic thromboembolic disease
Nonthrombotic embolization
Neoplastic emboli
Particulates and foreign material
Parasites
Chronic alveolar hypoxia
COPD
Interstitial lung disease
Hypoventilation syndromes
Pulmonary capillary hemangiomatosis
Left-sided cardiovascular disease
Mitral stenosis
Cor triatriatum
Aortic valvular disease
Cardiac tumors
Extrinsic pulmonary venous compression
Fibrosing mediastinitis
Pulmonary venoocclusive disease
TABLE 28.2 Updated Clinical Classification of Pulmonary Hypertension (2008 Dana Point Classification)
1.
Pulmonary arterial hypertension (PAH)
1.1
Idiopathic PAH
1.2
Heritable
1.2.1
Bone morphogenetic protein receptor type 2 germline mutations
1.2.2
Activin receptor-like kinase type 1 (endoglin) mutations, with or without hereditary hemorrhagic telangiectasia
Others: obstruction by neoplasm, fibrosing mediastinitis, chronic renal failure on dialysis
PULMONARY ARTERIAL HYPERTENSION
PAH is a syndrome that results from restriction of blood flow through the pulmonary arteries, leading to increased pulmonary vascular resistance and eventually to right heart failure. PAH usually can be attributed to an imbalance between vasodilating agents and vasoconstricting agents, with a relative paucity of prostacyclin and nitric oxide synthase expression and an increase in expression vasoconstrictive compounds, such as endothelin-1 and thromboxane A2. A relative procoagulant state is present, and inflammatory mediators, such as certain proinflammatory cytokines and autoantibodies, may also abound, suggesting that inflammation may play a role in the development of PAH in some patients.
The inciting factors are not always clearly known, but the pathological progression of PAH has been well characterized. Arterial medial hypertrophy, intimal proliferation and fibrosis, necrotizing arteritis, and plexiform lesions are manifestations of the progressive proliferation and destruction of the pulmonary arterial circulation. Superimposed organized thrombi may be present.
As outlined in Table 28-2, PAH consists of a number of subcategories, including idiopathic PAH (IPAH), heritable (familial PAH), and PAH associated with a number of conditions, including drugs and toxins and several systemic conditions.
Etiology and Pathogenesis of PAH
The prevalence of PAH is probably about 15 per million. IPAH is the most common form of PAH and affects females more commonly than males. IPAH represents patients with sporadic disease (no family history of pulmonary hypertension) and no identifiable risk factors. The term primary PAH, formerly in wide use, encompassed what is now considered IPAH, heritable PAH, and anorexigen-induced PAH. Heritable PAH (formerly referred to as familial PAH in the Avian-Venice/World Health Organization classifications of pulmonary hypertension) may be associated with a number of genetic derangements, including mutations in bone morphogenic protein receptor-2 and activin receptor-like kinase type 1, the latter most commonly coexisting with hereditary hemorrhagic telangiectasia. Heritable PAH accounts for less than 10% of cases of PAH. PAH is also associated with a number of other conditions, including exposures to certain drugs or toxins, connective tissue diseases, human immunodeficiency virus infection, congenital heart diseases associated with systemic-to-pulmonary shunts, portal hypertension, chronic hemolytic anemias, and schistosomaisis. Among the drug or toxic causes of PAH, potential etiologic agents are classified according to the strength of the evidence supporting their causal role in the development of PAH. The strength of the supporting evidence is graded as “definite,” “likely,” “possible,” or “unlikely.” A number of exposures potentially linked to the development of the strength of the evidence supporting their causal role in PAH are listed in Table 28-3.
PAH is associated with connective tissue diseases, but has only been well established in the setting of systemic sclerosis. Progressive systemic sclerosis produces changes in the vasculature more commonly than rheumatoid arthritis or systemic lupus erythematosus, and, therefore, has a higher association with pulmonary hypertension. The prevalence of pulmonary hypertension in patients with systemic sclerosis ranges from 4.9% to 26.7%. Pulmonary hypertension in patients with systemic sclerosis is a marker for poor prognosis. PAH is not the only cause of elevated pulmonary arterial pressure in patients with systemic sclerosis—obviously fibrotic lung disease may contribute to pulmonary hypertension and there may be an increased prevalence of diastolic dysfunction in these patients as well. It is important to note that PAH in patients with systemic sclerosis, and possibly other connective tissue diseases as well (particularly systemic lupus erythematosus, Sjögren’s syndrome, mixed connective disease, polymyositis, and rheumatoid arthritis), may occur independently of coexistent pulmonary and cardiac disease.
PAH is a rare complication of HIV infection and shares close similarities with IPAH in clinical, histopathological, and hemodynamic findings. The cause of PAH in patients with HIV infection is unknown, but humoral or indirect viral effects are suspected because the presence of viral DNA or the HIV virus itself has not been isolated from the pulmonary vascular endothelium.
TABLE 28.3 Associations with and Risk Factors for PAH
Untreated congenital heart defects, particularly those with left-to-right shunting, may develop PAH. Prolonged exposure of the pulmonary vasculature to increased blood flow and pressure results in arteriopathy, which eventually leads to persistently elevated pulmonary vascular resistance. The histopathological changes in PAH associated with congenital heart disease are similar to IPAH. Ultimately, pulmonary arterial pressure may rise sufficiently to produce shunt reversal and cyanosis—this situation is referred to as Eisenmenger’s syndrome.
PAH may be associated with portal hypertension— so-called portopulmonary hypertension. The prevalence of pulmonary hypertension in the setting of portal hypertension based on hemodynamic studies has been estimated at 2% to 6%. While liver disease is the primary condition associated with portal hypertension, the defining risk factor for portopulmonary hypertension is the presence of portal hypertension itself, not hepatic disease. The proposed mechanism for the development of pulmonary hypertension in patients with portal hypertension is thought to be the incomplete hepatic degradation of humoral factors that exert vasoconstricting and inflammatory effects on the pulmonary circulation. Two major risk factors are associated with the development of portopulmonary hypertension—female gender and autoimmune hepatitis. Other causes of elevated pulmonary arterial pressures are often present in patients with liver disease and portal hypertension, such as increased circulating fluid volume, a hyperdynamic circulatory state, and diastolic dysfunction, but, unlike portopulmonary hypertension, these conditions are uncommonly associated with elevated pulmonary vascular resistance. Because the causes of pulmonary hypertension in patients with portal hypertension may be mutlifactorial, right heart catheterization is required to establish the diagnosis of portopulmonary hypertension.
Only gold members can continue reading. Log In or Register to continue