radiosensitivity of a complex biologic system is determined by a number of variables. Some of these are inherent to the type of cells exposed while others relate to the cell’s current biochemical, mitotic, and oxygen tension status as well as many other variables at the time of irradiation. Damage observed at the molecular or cellular level may or may not result in clinically detectable adverse effects. Furthermore, although some responses to radiation exposure appear instantaneously or within minutes to hours, others take weeks, years, or even decades to appear.
electrons in motion, causing additional excitation and ionization along the path of the initial energetic electron. For example, a single 30 keV electron, set in motion following the photoelectric absorption of a single x-ray or γ-ray photon, can result in the production of over 1,000 low-energy secondary electrons (referred to as delta rays), each of which may cause additional excitation or ionization events in the tissue (Goodhead, 1994). This chain of ionizations ultimately gives rise to subexcitation electrons (i.e., electrons with kinetic energies less than the first excitation potential of liquid water, 7.4 eV) that become thermalized as they transfer their remaining kinetic energy by vibrational, rotational, and collisional energy exchanges with the water molecules. Observable effects such as chromosome breakage, cell death, oncogenic transformation, and acute radiation sickness, all have their origin in radiationinduced chemical changes in important biomolecules.
 ▪ FIGURE 20-1 A. Low-LET radiation like x-rays and γ-rays is considered sparsely ionizing on average; however, a majority of the radiation energy is deposited in small regions (on the scale of nanometers) via denser clusters of ionizations from low-energy secondary electrons. This illustration depicts primary and secondary electron tracks producing clusters of ionization events. The calculated number of tracks is based on a cell nucleus with a diameter of 8 µm. The track size is enlarged relative to the nucleus to illustrate the theoretical track structure. B. A segment of the electron track is illustrated utilizing a Monte Carlo simulation of clustered damage produced by ionizations and excitations along the path of a low-energy (300 eV) electron. Excitation and ionization along with secondary electrons are shown until the electron energy drops below the ionization potential of water (˜10 eV). C. DNA double helix drawn on the same scale as the ionization track. Complex clustered damage can result from closely spaced damage to the DNA sugar-phosphate backbone and bases from both direct ionizations and diffusion of OH radicals produced by the radiolysis of water molecules in close proximity (few nm) with the DNA (i.e., indirect effect). Multiple damaged sites are shown as green, or orange, explosion symbols that denote DNA strand breaks, or damaged bases, respectively. In this example, the result is a complex double-strand break, consisting of three strand breaks and three damaged bases, all within ten base pairs along the DNA. This type of complex DNA lesion is more difficult for the cell to repair and can lead to cell death, impaired cell function, or transformations with oncogenic potential. (Reprinted with permission from Goodhead DT. Energy deposition stochastics and track structure: what about the target? Radiat Prot Dosimetry. 2006;122(1-4):3-15. Copyright © Oxford University Press.) |
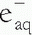 ). The
). The 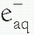 then reacts with another water molecule to form a negative water ion (H2O +
then reacts with another water molecule to form a negative water ion (H2O + 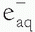 → H2O–). These water ions are very unstable; each rapidly forms another ion and a free radical:
→ H2O–). These water ions are very unstable; each rapidly forms another ion and a free radical: ▪ FIGURE 20-2 Physical and biologic responses to ionizing radiation. Ionizing radiation causes damage either directly by damaging the molecular target or indirectly by ionizing water, which in turn generates free radicals that attack molecular targets. The physical steps that lead to energy deposition and free radical formation occur within 10-5 to 10-6 s, whereas the biologic expression of the physical damage may occur from seconds to decades later. |

complexity of clustered DNA lesions) compared with low-LET radiation (e.g., x- and γ-rays). However, beyond approximately 100 keV/µm in tissue, the RBE decreases with increasing LET, because of the overkill effect. Overkill (or wasted dose) refers to the deposition of radiation energy in excess of that necessary to produce the maximal biologic effect. The RBE ranges from less than 1 to more than 20. For a particular type of radiation, the RBE depends on the biologic endpoint being studied. For example, chromosome aberrations, cataract formation, or acute lethality of test animals may be used as endpoints. Compared to high-energy γ-rays, the increased effectiveness of diagnostic x-rays in producing DNA damage is suggested not only by the differences in their microdosimetric energy deposition patterns but has also been demonstrated experimentally with an RBE of about 1.5 to 3. However, these differences do not necessarily imply (nor have epidemiological studies been able to confirm) an associated increase in cancer risk. The RBE also depends on the total dose, dose rate, fractionation, and cell type. Despite these limitations, the RBE is a useful radiobiologic tool that helps to characterize the potential damage from various types and energies of ionizing radiation. The RBE is an essential element in establishing the radiation weighting factors (wR) discussed in Chapter 3.
 ▪ FIGURE 20-3 The RBE of a given radiation is an empirically derived value that, in general (with all other factors being held constant), increases with the LET of the radiation. However, beyond approximately 100 keV/µm, the radiation becomes less efficient due to overkill (i.e., the maximal potential damage has already been reached), and the increase in LET beyond this point results in wasted dose. |
destruction of the cell by radiation but rather a radiation-induced loss of mitotic capacity (i.e., reproductive death). There is considerable evidence that damage to DNA (Fig. 20-4) is the primary cause of radiation-induced cell death.
 ▪ FIGURE 20-4 DNA is a primary target for damage that results in radiation-induced cell and tissue effects. The schematic illustrates the many orders of chromatin packaging from “naked” DNA to give rise to the highly condensed metaphase chromosome. The double helical DNA is wrapped around histones to form nucleosomes that are packaged to produce chromatin fibers that ultimately become highly packed in the chromosomes visible at mitosis. A mitotic chromosome is characterized by the centromere, which binds the two homologous chromatids together into the chromosome. The tips of each chromosome arm contain the telomeres. A gene is a sequence of nucleotides in a given position in the chromosome that is the functional unit of hereditary information, sometimes coding for a specific protein or controlling the function of other genetic material. (Courtesy of REAC/TS). |
the phosphate can rejoin, provided there is no opportunity for the broken portion of the strands to separate. While the rejoining is not typically immediate, because the broken ends require the action of a series of enzymes (endonuclease, polymerase, ligase) to rejoin, the rejoining is fast and the repair typically occurs with high fidelity. The presence of oxygen potentiates the damage by causing peroxidation of a base, which then undergoes radical transfer to the sugar, causing damage that prevents rejoining.
and germ cells and, if not repaired before DNA synthesis, may be transmitted during mitosis and meiosis. Chromosomal damage that occurs before DNA replication is referred to as chromosome aberrations, whereas that occurring after DNA synthesis is called chromatid aberrations. Unlike chromosomal aberrations, in chromatid aberrations, only one of the daughter cells will be affected if only one of the chromatids of a pair is damaged.
 ▪ FIGURE 20-5 A. Ionization patterns for low- and high-LET radiations in DNA. B. Types of radiation-induced DNA damages. (Reprinted with permission from Japan Atomic Energy Agency, Dependence of Yield of DNA Damage Refractory to Enzymatic Repair on Ionization & Excitation of Density Radiation, 2007. Copyright © JAEA.) |
arrest cell cycle progression) to allow for repair of damaged DNA or incompletely replicated chromosomes. In the case of potentially catastrophic DNA damage, the cell may initiate any of several cell death pathways (discussed below), effectively eliminating the damaged genetic material. The checkpoint and cell death responses (often known collectively as the DNA damage response [DDR]) utilize many of the same sensor molecules or complexes involved in DNA damage recognition and signal transduction. Many types of DNA repair mechanisms exist, including direct repair of a damaged nucleotide, base excision repair (BER), nucleotide excision repair (NER), SSB and DSB repair, and mismatch repair, each requiring its own set of enzymes (Fig. 20-6A). The repair of DNA damage depends on several factors, including the stage of the cell cycle and the type and location of the lesion.
 ▪ FIGURE 20-6 A. A summary of DNA repair processes. Each repair process is responsible for the repair of different types of DNA lesions. Some of the enzymes involved in each process are shown. Defects in some of these enzymes can lead to certain types of tumors or be targeted by certain drugs, as shown, for the treatment of cancers. (Reprinted with permission from Lord C, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287-294. Copyright © Springer Nature.) B. Scientists have recently been able to visualize the complicated and dynamic structures of DNA ligase using a combination of x-ray crystallography and small-angle x-ray scattering techniques. These experiments revealed the crystal structure of the human DNA ligase I protein bound to a short DNA oligonucleotide. The ring-shaped structure in the center of the figure is the solvent-accessible surface of the protein. The extended, chromosomal DNA (long coils) is an artist’s representation of the high-level organization of DNA structure. The figure illustrates the ring-shaped ligase protein sliding along the DNA searching for a break in the phosphodiester backbone of the DNA that is the substrate for the enzyme’s DNA end-joining activity. The enzyme, DNA ligase, repairs millions of DNA breaks generated during the normal course of a cell’s life, for example, linking together the abundant DNA fragments formed during replication of the genetic material in dividing cells. DNA ligase switches from an open, extended shape to a closed, circular shape as it joins DNA strands together. (Courtesy of Tom Ellenberger, DVM, PhD, Department of Biochemistry and Molecular Biophysics at Washington University School of Medicine, St. Louis, MO.) C. DSB induction and repair in primary human fibroblasts. Using immunofluorescence techniques, a fluorescent antibody-specific for γ-H2AX (a phosphorylated histone) forms discrete nuclear foci that can be visualized at sites of DSBs. DSB repair was evaluated at 3 min and 24 h after exposure to 200 mGy; repair was almost complete at 24 h. The length of the white scale bar shown in the unirradiated control panel equals 10 µm. (Reprinted with permission from Rothkamm K, Löbrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A. 2003;100:5057-5062. Copyright © National Academy of Sciences.) |
 ▪ FIGURE 20-7 Comparison of the two pathways for repair of DNA double-strand breaks: homologous recombination (HR) and non-homologous end-joining (NHEJ). Because HR usually uses the sister chromatid for the repair, it can only occur in the late S or G2 phases of the cell cycle, after DNA replication, but it is a highly accurate repair. On the other hand, the more common repair process, NHEJ, can occur at any time in the cell cycle but is an error-prone process that ligates the broken ends of DNA together, often resulting in loss of genetic information. |
400 mGy for chronic exposure can be detected with confidence limits that do not include zero. Although many chromosomal aberrations are unstable and gradually lost from circulation, this assay is generally considered the most sensitive method for estimating recent exposure (i.e., within 6 months). More persistent, stable reciprocal translocations can be measured using fluorescence in situ hybridization (FISH). In this method, chromosomes are labeled with chromosome-specific fluorescent DNA probes, allowing translocations to be identified using fluorescent microscopy (Fig. 20-9E). Reciprocal translocations are believed to persist for a considerable period after the exposure, and this approach has been used as one of the methods to estimate the doses to survivors of the atomic bombs detonated in Hiroshima and Nagasaki decades ago.
 ▪ FIGURE 20-8 Model of the key steps required for NHEJ and HR repair of DNA DSBs. (Reprinted with permission from Chowdhury D, Choi Y, Brault M. Charity begins at home: non-coding RNA functions in DNA repair. Nat Rev Mol Cell Biol. 2013;14:181-189. Copyright © Springer Nature.) |
 ▪ FIGURE 20-9 Examples of chromosomal aberrations and the effect of recombinations, replication, and anaphasic separation. A. A single break in one chromosome, which results in centric and acentric fragments. The acentric fragments are unable to attach to the mitotic spindle and are transmitted to only one of the daughter cells where they may remain in the cytoplasm. These fragments are eventually lost in subsequent divisions. B. Ring formation may result from two breaks in the same chromosome in which the two broken ends of the centric fragment recombine. The ring-shaped chromosome undergoes normal replication, and the two (ringshaped) sister chromatids separate normally at anaphase—unless the centric fragment twists before recombination, in which case the sister chromatids will be interlocked and unable to separate. C. Translocation may occur when two chromosomes break and the acentric fragment of one chromosome combines with the centric fragment of the other and vice versa, or (D) the two centric fragments recombine with each other at their broken ends, resulting in the production of a dicentric. E. Metaphase spread, containing a simple dicentric interchange between chromosomes 2 and 8 visualized with multiplex fluorescence in situ hybridization (mFISH). This technique utilizes fluorescently labeled DNA probes with markers specific to regions of particular chromosomes, which allows for the identification of each homologous chromosome pair by its own color. The color is computer generated based on differences in fluorescence wavelength among probes. (Reprinted with permission from Cornforth MN, et al. Chromosomes are predominantly located randomly with respect to each other in interphase human cells. J Cell Biol. 2002;159(2):237-244. Copyright © Rockefeller University Press.) |
in the cell cycle at the time of exposure) as well as a number of physical factors related to the radiation exposure (e.g., dose, dose rate, LET), a number of responses are possible such as delayed cell division, apoptosis, reproductive failure, genomic instability (delay expression of radiation damage), DNA mutations including phenotypic (including potentially oncogenic) transformations, bystander effects (damage to neighboring unirradiated cells), and adaptive responses (irradiated cells become more radioresistant). Many of these effects are discussed in more detail below. While a wide variety of the biologic responses to radiation have been identified, the study of radiation-induced reproductive failure (also referred to as clonogenic cell death or loss of reproductive integrity) is particularly useful in assessing the relative biologic impact of various types of radiation and exposure conditions. The use of reproductive integrity as a biologic effects marker is somewhat limited, however, in that it is applicable only to proliferating cell systems (e.g., stem cells). For differentiated cells that no longer have the capacity for cell division (e.g., muscle and nerve cells), cell death is often defined as loss of specific metabolic functions or functional capacity. One must also keep in mind that, with many of the assays described below, sensitivity to detect changes may be limited at low radiation doses and dose rates, so data obtained at higher doses are often back-extrapolated, using various mathematical models discussed below, to low doses and dose rates.
Sublethal damage is a concept based on experiments that show that when the radiation dose is split into two or more fractions, with sufficient time between fractions, the cell survival increases after low-LET radiation. The presence of the shoulder in a cell survival curve is taken to indicate that more than one ionizing event (“hit”), on average, is required to kill a cell and the reappearance of the shoulder when a large dose is delivered in fractions indicates that the cells are capable of repairing sublethal damage between fractions.
 ▪ FIGURE 20-10 Typical cell survival curve illustrating the portions of the curve used to derive the extrapolation number (n), the quasithreshold dose (Dq), and the D0 dose. |
The LQ (or alpha-beta model, as many call it) is more commonly used than the previously described n–D0 model, for several reasons: the LQ model is mechanistically based, it is more useful in radiotherapy for explaining fractionation effect differences between late responding normal tissues and early responding tissues or tumors, and the LQ model seems to fit most experimental data on human cell lines. The dose at which cell killing is equal from the linear (αD) and quadratic (βD2) contributions is referred to as the α/β ratio. The α/β ratio is a measure of the curvature of the cell survival curve and, thus, a measure of the sensitivity of different cell types to fractionation of radiation dose, Figure 20-11. For example, late responding normal tissues such as spinal cord or lung that have smaller α/β ratios of 3 or 4 are preferentially “spared” by fractionation compared to tumors and early responding normal tissues (gut, skin, bone marrow) where the α/β ratio is larger (8 to 12), indicating less ability to repair (i.e., more alpha component and less effect of fractionating the dose).
bodies composed of cytoplasm and tightly packed organelles that are eliminated by phagocytosis. Hallmarks of apoptosis include the sequential activation of caspases (cysteine-dependent aspartate-directed proteases) from pro-caspases; interactions of pro- and anti-apoptotic members of the bcl-2 family of proteins, many working at the level of the mitochondria; cleavage of multiple proteins; and, ultimately, cleavage of DNA between nucleosomes to form characteristic fragments consisting of multiples of the amount of DNA in a nucleosome. Extrinsic apoptosis is initiated at the cell surface with activation of death receptors such as CD95 (Fas) receptor, dimerization and activation of the initiator, or upstream caspase, caspase-8, which, in turn, activates the downstream caspase-3, which activates endonucleases and other proteases
to cleave DNA and many other cellular proteins. Intrinsic apoptosis is generally started at mitochondria where interactions of pro-apoptotic proteins, such as Bax and Bak, with anti-apoptotic Bcl-2 and Bcl-XL results in the release of cytochrome c from the mitochondria, formation of apoptosomes, activation of caspase-9, activation of caspase-3, and the cleavage of other cellular proteins and DNA. Radiation can induce apoptosis through DNA damage initiating the formation of pro-apoptotic proteins such as Noxa and Puma, which activate intrinsic apoptosis or upregulation of death receptors to begin extrinsic apoptosis pathways. This is an over-simplistic description of the processes, as there can be much cross-talk among pathways and regulation by other proteins, for example, p53 or XIAP (sex-linked inhibitor of apoptosis) at various steps.
 ▪ FIGURE 20-11 The LQ model. The experimental data are fitted to a LQ function. There are two components to cell killing: One is proportional to dose (αD); the other is proportional to the square of the dose βD2. The dose at which the linear and quadratic components are equal is the ratio α/β. The LQ curve bends continuously but is a good fit to the experimental data for the first few decades of survival. (Reprinted with permission from Hall EJ, Giaccia AJ. Radiobiology for the Radiologist. 7th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2012.) |
 ▪ FIGURE 20-12 Comparison of morphological changes in cells undergoing three different modes of radiationinduced cell death—apoptosis, autophagy, and necrosis. A particular mode of cell death may predominate depending on cell type, radiation quality, and dose, and other environmental factors. (Reprinted with permission from Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570-1583.) |
 ▪ FIGURE 20-13 Cascades of molecular events involved in apoptosis (A), autophagy (B), and necroptosis (C). See text for explanations. (A and C: Reprinted with permission from Matt S, Hofmann TG. The DNA damageinduced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. 2016;73:2829-2850. B: Reprinted from Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570-1583.) |
 ▪ FIGURE 20-14 Cell survival curves illustrating the effect of dose rate for low-LET radiation. Lethality is reduced because the repair of sublethal damage is enhanced when a given dose of radiation is delivered at a low versus a high dose rate. |
 ▪ FIGURE 20-15 Cell survival curves illustrating the greater damage produced by radiation with high-LET. At 10% survival, high-LET neutron radiation is three times as effective as the same dose of low-LET radiation in this example. |
fractions of 2 Gy with sufficient time between fractions for repair of sublethal damage. For low-LET radiation, the decreasing slope of the survival curve with decreasing dose rate (see Fig. 20-14) and the reoccurrence of the shoulder with fractionation (see Fig. 20-16) are clear evidence of repair.
 ▪ FIGURE 20-16 Idealized fractionation experiment depicting the survival of a population of 106 cells as a function of dose. Curve A represents one fraction of 10 Gy. Curve F represents the same total dose as in curve A delivered in equal fractionated doses (D1 through D5) of 2 Gy each, with intervals between fractions sufficient to allow for repair of sublethal damage. (Modified from Hall EJ. Radiobiology for the Radiologist. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2000.) |
extremely radiosensitive compared with other cells in the body. On the other end of the spectrum, the fixed postmitotic neurons found in the central nervous system (CNS) are relatively radioresistant (Fig. 20-18). This classification scheme was refined in 1968 by Rubin and Casarett, who defined five cell types according to characteristics that affect their radiosensitivity (Table 20-1).
 ▪ FIGURE 20-17 Cell survival curves demonstrating the effect of oxygen during high (blue) and low (green) oxygen tension on the OER for high- and low-LET irradiation. |
phase because when it is prevented by genetic alterations or drugs such as caffeine, cells are sensitized to radiation. Also important for radiation sensitivity is the ability to arrest in the G1 phase, which is highly dependent on cells having a functional p53 pathway (see bottom of Fig. 20-19). The tumor suppressor gene TP53 (so named because it encodes a phosphorylated protein with a molecular weight of 53 kDa) operates predominantly at the G1/S checkpoint. The p53 protein (discussed again in relation to radiation-induced carcinogenesis later in the chapter) induces cellcycle arrest through the up-regulation of cyclin-dependent kinase inhibitors and thus allows for repair of DNA damage. The CIP/KIP (CDK interacting protein/Kinase inhibitory protein) family is one of two families (CIP/KIP and INK4) of mammalian cyclin-dependent kinase (CDK) inhibitors (CKIs) involved in regulating the cell cycle. The CIP/KIP family members also have a number of CDK-independent roles involving regulation of transcription, apoptosis, and the control of the cell’s cytoskeleton. Thus the p53 protein activation of cyclin-dependent kinases can activate DNA repair mechanisms or, in the case of severe DNA damage, induce cell death via apoptosis.
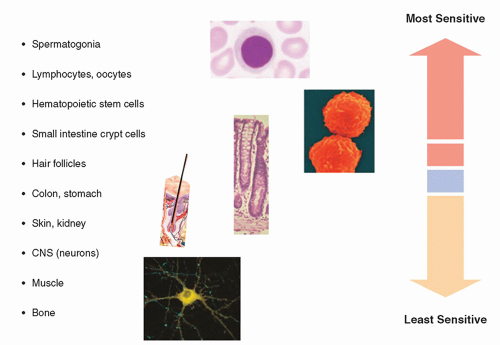 ▪ FIGURE 20-18 Relative radiosensitivity of tissues. |
TABLE 20-1 CLASSIFICATION OF CELLULAR RADIOSENSITIVITY | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
dose and dose rate as well as among lymphocytes from different individuals and with other variables. Many other endpoints for adaptive response have been studied such as cell lethality, mutations, and defects in embryonic development for which the evidence for an adaptive response was highly variable. While many theories have been advanced to explain this phenomenon, there is still insufficient evidence to use these results to modify the dose-response relationship for human exposure to radiation.
 ▪ FIGURE 20-19 Phases of the cell’s reproductive cycle. The time between cell divisions is called interphase. Interphase includes the period after mitosis but before DNA synthesis (G1), which is the most variable in length of the phases; followed by S phase, during which DNA synthesis occurs; followed by G2, all leading up to mitosis (M phase), the events of which are differentiated into prophase, metaphase, anaphase, and telophase. Much of the control of the progression through the phases of a cell cycle is exerted by specific cell cycle control genes at checkpoints. Checkpoints are critical control points in the cell cycle that have built-in stop signals that halt the cell cycle until overridden by external chemical signals to proceed. There are three major checkpoints in the cell cycle, G1 checkpoint between G1 and S phase (G1/S), the G2 checkpoint between G2 and mitosis (G2/M), and the M (metaphase) checkpoint. The G1/S checkpoint is where the cell monitors its size, available nutrients, and the integrity of DNA (e.g., prevents copying of damaged bases, which would fix mutations in the genome) to assure all are adequate for DNA synthesis and progression on through the cell division cycle. In the absence of a proceed signal, the cell will enter a quiescent state G0 (the state of most cells in the body) until it receives a stimulation signal to continue. Following DNA synthesis, the G2/M checkpoint occurs at the end of the G2 phase, during which DNA damage induced during replication, such as mismatched bases and double-stranded breaks, is repaired. The cell ensures that DNA synthesis (S-phase) has been successfully completed before triggering the start of mitosis, and at the metaphase (spindle) checkpoint the cell monitors spindle formation and ensures all chromosomes are attached to the mitotic spindle by kinetochores prior to advancing to anaphase. p53, the product of the tumor suppressor gene TP53, operates predominantly at the G1/S checkpoint. The p53 pathway (inset) is vital to maintaining cell health, monitoring incoming stress from various sources such as oxidative stress, hypoxia, and DNA damage to name a few. Depending on the stressor, the response of the p53 pathway will change leading to cell cycle arrest, apoptosis, senescence, DNA repair, and metabolism adjustment. |
factors to predict the biological consequences of radiation exposure in humans. On the other hand, the nonlinear nature of these and other multicellular and tissue-level responses raises serious questions regarding the current paradigm of linear extrapolation of risk based on the individual cell and the target. An active area of current research focused on addressing these complex responses to radiation interactions is a multidimensional, systems-level approach that includes the integration of radiation epidemiology with radiobiological investigations.
group of soluble short-acting proteins, glycoproteins, and peptides produced by various immune and vascular cells that activate specific receptors and modulate the functions of many cells and tissues. Some cytokines may be membrane-bound or associated with ECM. Cytokines released by the vascular endothelium of irradiated tissues are implicated in the acute phase response to ionizing radiation and other inflammatory stimuli. Examples of radiation-induced cytokines include tumor necrosis factor (TNF3)-α, Interleukin (IL)-1, transforming growth factor (TGF)-β, and stem cell factor. Although many of these cytokines and growth factors, when induced by radiation, increase tissue damage, for example, the role of TGF-β in pneumonitis
is well documented, some growth factors can be radioprotective in some tissues, e.g., basic fibroblast growth factor protects microvasculature and IL-1 is a radioprotector of hematopoietic cells (Hall and Giaccia, 2018).
TABLE 20-2 RUBIN AND CASARETT CLASSIFICATION OF RELATIVE ORGAN AND TISSUE RADIOSENSITIVITY AND PRIMARY MECHANISM FOR RADIATION-INDUCED PARENCHYMAL HYPOPLASIA | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
 ▪ FIGURE 20-20 Histopathology showing radiation-induced arteriole fibrosis (left). (From Zaharia M, Goans RE, Berger ME, et al. Industrial radiography accident at the Yanango hydroelectric power plant. In: Ricks RC, et al., eds. The Medical Basis for Radiation Accident Preparedness, the Clinical Care of Victims. New York, NY: The Parthenon Publishing Group; 2001:267-281.) |
 ▪ FIGURE 20-21 Schematic diagram of organ system response to radiation. |
seen. At higher doses, radiation can interfere with normal maturation, reproduction, and repopulation of germinative epidermal cell populations. At very high doses, the mitotic activity in the germinal cells of the sebaceous glands, hair follicles, basal cell layer, and intimal cells of the microvasculature can be compromised.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 ▪ FIGURE 20-22 Examples of radiation-induced effects on skin. A. National Cancer Institute (NCI) skin toxicity grade 1: Two fluoroscopically guided procedures were performed through overlapping skin ports in a 65-year-old man. Note enhanced reaction in the overlap zone. The first procedure was performed 6 weeks before and the second procedure, 2 weeks before this photograph was obtained (From Balter S, Hopewell JW, Miller DL, et al. Fluoroscopically guided interventional procedures: a review of radiation effects on patients’ skin and hair. Radiology. 2010;254(2):326-341). B. Technologist error resulted in a 2-year-old child being accidentally exposed to radiation from 151 CT slice acquisitions without the table indexing and thus all were through almost the same 3 mm tissue plane. Within several hours after the failed CT scan, a line of erythema developed across the patient’s face in the same distribution (arrows). Peak skin and brain dose within the slice were estimated to be 7.2 Gy and 5.2 Gy, respectively. CT scan parameters were set at 300 mAs and 120 kV. (Photo courtesy of Dr. Fred Mettler, dose estimates courtesy of Drs. Jerrold T. Bushberg and J. Anthony Seibert.). C. Patient with temporary hair loss in the region of four MDCT perfusion studies and two angiographies of the head within 15 days of admission for suspected stroke. Epilation appeared on day 37 after the first perfusion study and lasted for 51 days. Peak skin dose estimates for each of the MDCT procedures was ˜1.93 Gy. (Reprinted with permission from Imanishi Y, Fukui A, Niimi H., et al. Radiation-induced temporary hair loss as a radiation damage only occurring in patients who had the combination of MDCT and DSA. Eur Radiol. 2005;15:41-46. Copyright © Springer Nature.) D. A 62-year-old man with a history of 2 previous cardiac catheterizations approximately 5 years prior. Lesion that had been developing for over a year presents as an NCI skin toxicity grade 3 chronic radiodermatitis at the site of beam entry. The lesion is an 8″ × 6″ well-demarcated erythematous atrophic plaque with telangiectasias and ulceration. (Reprinted with permission from Spiker, A et al. Fluoroscopy-induced chronic radiation dermatitis. AJR 2012:1861-1863. Copyright © Elsevier.) E. Dry desquamation (poikiloderma) at one month in a patient receiving approximately 11 Gy calculated peak skin dose. (Reprinted with permission from Chambers C, Fetterly K, Holzer R, et al. Radiation safety program for the cardiac catheterization laboratory. Catheter Cardiovasc Interv. 2011;77. Copyright © Wiley.). F-H. NCI skin toxicity grade 4. A 40-year-old male who underwent multiple coronary angiography and angioplasty procedures. The photographs show the time sequence of a major radiation injury. (Reprinted with permission from Shope TB. Radiation-induced skin injuries from fluoroscopy. RadioGraphics 1996;16:1195-1199. Copyright © Radiological Society of North America.) (F) Six to eight weeks postexposure (prolonged erythema with a mauve central area, suggestive of ischemia). The injury was described as “turning red about 1 month after the procedure and peeling a week later.” By 6 weeks, it had the appearance of a second-degree burn; (G) sixteen to twenty-one weeks postexposure (depigmented skin with a central area of necrosis); and (H) eighteen to twenty-one months postexposure (deep necrosis with atrophic borders). Skin breakdown continued over the following months with progressive necrosis. The injury eventually required a skin graft. While the magnitude of the skin dose received by this patient is not known, from the nature of the injury it is probable that the dose exceeded 20 Gy. This sequence is available on the FDA Web site. (National Council on Radiation Protection and Measurements. Radiation Dose Management for Fluoroscopically-Guided Interventional Procedures. NCRP Report No. 168. Bethesda, MD: National Council on Radiation Protection; 2010.) |
 ▪ FIGURE 20-22 (Continued) I. A three-dimensional view depicting the spectrum of radiation-induced effects on skin as shown in the previous photos (A-H) and discussed in the text. (Courtesy of Nicholas Zaorsky, MD.) |
duration of temporary sterility is dose dependent, with recovery beginning at 1 and as long as 3.5 years after doses of 1 and 2 Gy, respectively. However, following exposure (and provided the dose is not excessive), there will be a window of fertility before the onset of sterility, as long as mature sperm are available. Chronic exposures of 20 to 50 mGy/wk can result in permanent sterility when the total dose exceeds 2.5 to 3 Gy. The reduced threshold for effect following chronic versus acute exposure is unusual (i.e., an inverse fractionation effect) and is believed to be due to stem cells progressing into radiosensitive stages (Lushbaugh and Ricks, 1972). Reduced fertility due to decreased sperm count (oligospermia) and motility (asthenozoospermia) can occur 6 weeks after a dose of 150 mGy. These effects are not related to diagnostic examinations, because acute gonadal doses exceeding 100 mGy are unlikely.
TABLE 20-4 PHYSICAL AND BIOLOGICAL MODIFIERS OF RADIATION-INDUCED SKIN DAMAGE | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
provided the exposure is not so high as to destroy the relatively radioresistant small primordial follicles. The dose that will produce permanent sterility is age dependent, with higher doses (˜10 Gy) required to produce sterility prior to puberty than in premenopausal women over 40 years old (˜2 to 3 Gy).
mSv/y to the lens of the eye may need to be reevaluated. However, the proposed ICRP limit is almost a factor of 10 lower than current limits and lower than the whole-body dose limit in the United States of 50 mSv/y. Similarly, the NCRP has recommended an occupational dose limit for the lens of the eye of 50 mGy/y (NCRP, 2016), although the U.S. national regulations have not yet been revised. Adoption of ICRP or NCRP recommendations by regulatory bodies would present new challenges for radiation protection in health care settings, especially for those involved in performing fluoroscopically guided interventional procedures. In any case, the use of eye protection in the form of leaded glasses and/or ceiling mounted lead acrylic shielding is imperative for workers whose careers will involve long-term exposure to scattered radiation.
approximately 3 weeks after the poisoning from the complications of profound pancytopenia that is characteristic of severe hematopoietic damage.
TABLE 20-5 THRESHOLD DOSES IN TISSUES AND ORGANS IN ADULTS EXPOSED TO ACUTE, FRACTIONATED OR PROTRACTED, AND CHRONIC RADIATION EXPOSUREa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
for approximately 2 to 4 weeks or in some cases even longer. This stage is the most difficult to manage from a therapeutic standpoint, because of the overlying immunoincompetence that results from damage to the hematopoietic system. Therefore, treatment during the first 6 to 8 weeks after the exposure is essential to optimize the chances for recovery. If the patient survives the manifest illness stage, recovery is likely; however, the patient will be at higher risk for cancer and, to a much lesser extent, his or her future progeny may have an increased risk of genetic abnormalities.
 ▪ FIGURE 20-23 ARS follows a clinical pattern that can be divided into three phases: (1) an initial or prodromal phase that presents as non-specific clinical symptoms, such as nausea, vomiting, and lethargy (hematological changes may also occur during this period); (2) the latent phase, during which the prodromal symptoms typically subside; and (3) the manifest illness phase, during which the underlying organ system damage is expressed. The type, time of onset, and severity of prodromal symptoms are dose dependent. The duration of the latent period, as well as the time of onset and severity of the manifest illness phase, and ultimate outcome are all, to a variable extent, dependent upon total dose, uniformity of the exposure, and individual radiation sensitivity. As a rule, higher doses shorten the time of onset and duration of all three phases and increase the severity of the prodromal and the manifest illness phases. |
TABLE 20-6 CLINICAL FINDINGS DURING PRODROMAL PHASE OF ARS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:
 X-ray Production, Tubes, and Generators
X-ray Production, Tubes, and Generators
 X-ray Dosimetry in Projection Imaging and Computed Tomography
X-ray Dosimetry in Projection Imaging and Computed Tomography
 Radiation Detection and Measurement
Radiation Detection and Measurement
 Magnetic Resonance Basics: Magnetic Fields, Nuclear Magnetic Characteristics, Tissue Contrast, Image Acquisition
Magnetic Resonance Basics: Magnetic Fields, Nuclear Magnetic Characteristics, Tissue Contrast, Image Acquisition
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


