Ashwin Singh Parihar1,2 and Erik Mittra3 1 Department of Nuclear Medicine, Postgraduate Institute of Medical Education and Research, Chandigarh, India 2 Mallinckrodt Institute of Radiology, Washington University School of Medicine, St Louis, MO, USA 3 Department of Diagnostic Radiology, Division of Nuclear Medicine & Molecular Imaging, Oregon Health & Science University, Portland, OR, USA Radionuclide therapies are currently expanding rapidly due to a variety of factors, including novel targets, their proven efficacy and safety, and the concept of theranostics. The latter is a portmanteau of diagnostics and therapeutics. Accurate noninvasive visualization, using diagnostic molecular targets for functional imaging, is a critical tool for staging, restaging, and response assessment for a large number of cancers. Using those same molecular targets to deliver precision radionuclide therapy is the underlying principle of theranostics in nuclear medicine. Initially used for the treatment of hyperthyroidism and differentiated thyroid malignancies using 131I, the concept of theranostics has since grown to cover a diverse range of malignancies, such as those of prostate, liver, neuroendocrine neoplasms (NENs), painful osseous metastases, and hematologic malignancies, among others. In this chapter, we review the principal radionuclide therapies with regards to their clinical applications, correlative functional imaging, pre‐ and post‐therapeutic considerations, and efficacy and safety. NENs, originating from the secretory cells of the neuroendocrine system, have an endodermal lineage. These heterogeneous tumors, with widely varying grades and degrees of differentiation and ultimately prognosis, are classified according to their site of origin (e.g. bronchopulmonary, gastro‐entero‐pancreatic (GEP), breast, thyroid, uterus, and others) [1]. The most common among these are the GEP NENs. Based on the embryonic origin, the gastrointestinal (GI) NENs are further classified into foregut (stomach, duodenum), midgut (jejunum, ileum, cecum, appendix, proximal colon), and hindgut (distal colon, rectum) tumors [2]. Midgut NENs are the most common and, by secretion of vasoactive hormones, most frequently lead to the functional tumors or the characteristic “carcinoid syndrome” constituted by flushing, diarrhea, and, less frequently, right‐sided valvular heart disease [3]. NENs are known to abundantly express somatostatin receptors (SSTRs), which have a role in modulating the proliferation and secretory activity of the tumor cells. This has therapeutic implications, as the somatostatin analogues such as octreotide and lanreotide are used for achieving symptomatic control in patients with functional tumors and have antiproliferative properties [4, 5]. SSTR expression has also been utilized for functional imaging, initially for gamma scintigraphy with diethylenetriamine pentaacetic acid (DTPA)‐octreotide (pentetreotide) then onto positron emission tomography (PET) with the 1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid (DOTA) peptides (DOTA‐[1‐Nal3]octreotide, DOTANOC; DOTA‐[Tyr3]octreotide, DOTATOC; DOTA‐[Tyr3]octreotate, DOTATATE) [6]. Out of the five types of SSTRs (SSTR1–5), SSTR2 forms the predominant receptor in the GEP NENs and is bound by all these receptor agonists. DOTANOC has demonstrated high affinity and a broader spectrum of binding to SSTR2, ‐3, and ‐5 in comparison to its counterparts [7]. Development of peptide receptor radionuclide therapy (PRRT) is also based on targeting the same SSTR. 111In‐pentetreotide was used in initial trials, albeit with limited success due to the physical characteristics of the auger electron emissions of this primary gamma‐emitter [8]. As newer analogues were developed that could be attached to beta‐emitting radionuclides such as 90Y and 177Lu, the efficacy of PRRT also improved significantly [9]. Based on the results of the NETTER‐1 trial [10] and a study from Netherlands [11], Lutathera (177Lu‐DOTATATE, Advanced Accelerator Applications Inc., USA) was the first (and so far only) approved PRRT radiopharmaceutical for GEP NENs by the United States Food and Drug Administration (US FDA) in 2018. Therefore, the remainder of this section will focus on 177Lu‐DOTATATE‐based PRRT. There are, however, ongoing studies exploring the use of PRRT using other agents such as receptor antagonists with higher binding activity to the SSTR or labeled with alpha‐emitters such as 225Ac and 213Bi, with initial reports showing promising results [12, 13]. Functional imaging with radiolabeled somatostatin analogues is useful both from a purely diagnostic perspective and as an adjunct to PRRT (selecting patients and assessing response) (Figures 32.1 and 32.2). Presently, the DOTA‐peptides mentioned above are typically labeled with 68Ga for PET‐based SSTR imaging of NENs in diagnostic and theranostic settings [14]. DOTATATE is also available labeled with 64Cu, which has the advantages of not requiring a generator, a much longer half‐life, and a shorter positron range than 68Ga, but the disadvantage of a lower photon percentage. A list of practical indications of SSTR imaging is provided in Table 32.1. Figure 32.1 Checklist for PRRT planning. Figure 32.2 63‐year‐old male with a pancreatic neuroendocrine neoplasm with liver metastases previously treated with a pancreaticoduodenectomy, adjuvant carboplatin, and liver‐directed therapy. Due to continued progression of disease, he was referred for . 68Ga‐DOTATATE PET/CT [maximum intensity projection image (a), axial CT (b), PET (c), and fusion PET/CT (d)] shows extensive SSTR‐expressing metastatic disease, with maximal avidity higher than that of spleen. Based on this, he started PRRT. Planar post‐therapy scintigraphy image (e) shows 177Lu‐DOTATATE accumulation in the same distribution as seen on the pre‐therapy PET images, albeit with lower resolution due to gamma‐imaging. Table 32.1 Present and evolving indications of functional imaging with somatostatin receptor analogues PRRT with 177Lu‐DOTATATE requires careful patient screening with the assessment of appropriateness by a dedicated multidisciplinary team, including medical and surgical oncology, nuclear medicine, diagnostic and interventional radiology, endocrinology, and pathology. The individualized goals of therapy (Table 32.2) have to be clearly identified during the patient screening procedure. The initial screening of a patient for PRRT includes review of the tumor histopathology. Tumor grade (assessed by mitotic activity or Ki‐67 labeling index; Table 32.3) and degree of differentiation are two key histologic parameters affecting choice of management and disease prognosis. While tumor grade denotes the proliferative activity of the tumor cells, degree of differentiation assesses their resemblance to the normal counterparts [1]. Patients with G1 or G2 tumors that are well‐differentiated are typically suitable candidates for PRRT. Table 32.2 Goals of PRRT in different clinical settings Table 32.3 Tumor grade assessed by mitotic activity and Ki‐67 labeling index Source: Based on Klimstra et al. [15]. a 2 mm2 denotes 10 high power fields at 40× magnification with a field diameter of 0.5 mm. 111In‐pentreotide and 99mTc‐hydrazinonicotinamide (HYNIC)‐TOC based single‐photon emission computed tomography (SPECT) imaging or 68Ga‐ or 64Cu‐labeled somatostatin‐analogue based PET imaging is performed to noninvasively document SSTR expression on tumor cells [16]. PET is favored over SPECT due to its higher sensitivity, better radiotracer kinetics, resolution, and dosimetry [17]. Deoxy‐2‐[18F]fluoroglucose (18F‐FDG) PET/CT, when performed pretherapy, can provide additional useful prognostic information, especially for those with higher‐grade or more poorly differentiated tumors [18] (Figure 32.3). The laboratory investigations that have to be performed for patient selection are provided in Table 32.4 with the reference cutoff values [19]. Figure 32.1 provides a checklist for planning PRRT with 177Lu‐DOTATATE. Figure 32.3 63‐year‐old female with biopsy proven metastatic right axillary neuroendocrine tumor underwent 68Ga‐DOTATATE PET/CT which showed somatostatin receptor expressing (SUVmax 5.9) left axillary lesion (A, thick arrow) and a faintly somatostatin receptor expressing (SUVmax 2.6) soft tissue lesion in the posterior basal segment of the left lung lower lobe (not seen on the maximum intensity projection image). Both lesions showed higher avidity on 18F‐FDG PET/CT (B, axillary lesion [SUVmax 7.4], thick arrow; lung lesion [SUVmax 4.1], thin arrow), as expected of a higher‐grade tumor. Table 32.4 Laboratory investigations prior to first cycle of PRRT with 177Lu‐DOTATATE Source: Modified from Hope et al. [19]. 177Lu‐DOTATATE, diluted in normal saline with an activity of 7.4 GBq (200 mCi), is administered intravenously over 30 minutes during each cycle of therapy. A running intravenous line is preferred for infusing the radiopharmaceutical as this minimizes the risk of extravasation. Appropriate radiation shielding has to be kept in place to avoid extra radiation burden to staff. Care should also be taken to avoid spillage and contamination. Table 32.5 Requirements of amino acid formulation for 177Lu‐DOTATATE‐based PRRT Source: Modified from Hope et al. [19]. The patient should be monitored for any adverse events during the therapy and availability of octreotide, and corticosteroids and intravenous fluids should be ensured to manage carcinoid crisis in the rare event that it occurs. The patient can be discharged after clinical assessment and in line with local radiation safety protocols. A post‐therapy whole‐body scintigraphy (with or without SPECT), performed immediately after or within 48 hours after the therapy, helps in confirmation of radiotracer uptake at the involved sites. Since significant radionuclide excretion occurs via urine, and precautions to avoid urinary contamination have to be explained to the patients and their care‐givers. Post‐therapy, patients require laboratory investigations (complete blood count, liver and renal function tests) at 4 (±2) and 8 weeks. Typically, the interval between each cycle of therapy is 8 weeks with a total of four cycles. Diagnostic imaging for response assessment should be done at 1–3 months (may be performed at the end of two cycles if clinically indicated), 6 months, and 12 months after completion of all cycles. National Comprehensive Cancer Network (NCCN) guidelines recommend contrast‐enhanced abdominal and pelvic CT or contrast‐enhanced magnetic resonance imaging (MRI) with chest CT for diagnostic imaging to assess treatment response [21]. The role of SSTR‐based functional imaging for response assessment is still being evaluated but can be done as an adjunct to conventional imaging, especially when that is ambiguous. PRRT with 177Lu‐DOTATATE has been proven to have good efficacy in NENs in multiple studies across various clinically diverse settings. The NETTER‐1 phase 3 trial in patients with midgut NEN compared 177Lu‐DOTATATE with 30 mg octreotide long‐acting repeatable (LAR) in the test group versus octreotide LAR 60 mg alone in the control group. The test group had a significantly higher progression‐free survival rate of 65.2% (95% confidence interval [CI] 50.0–76.8) at 20 months versus 10.8% (95% CI 3.5–23.0) in the control group. The test group also had a significantly higher treatment response rate of 18% versus 3% in the control group. The median progression‐free survival was not reached in the test group and was 8.4 months (95% CI 5.8–9.1) in the control group [10]. Similar results were shown by a retrospective study including the Dutch population, in which an overall best objective response rate (including complete and partial response [, as per RECIST 1.1) of 39% was seen with 177Lu‐DOTATATE therapy in a total of 443 patients with different NENs. The median overall and progression‐free survivals were 63 and 29 months, respectively [11]. Based on the findings of a systematic review and network meta‐analysis of multiple randomized control trials (RCTs), the combination therapy of 177Lu‐DOTATATE with a somatostatin analogue showed the second highest treatment efficacy in GI NENs, exceeded only by combination therapy with bevacizumab and a somatostatin analogue. Furthermore, combination therapy of 177Lu‐DOTATATE with a somatostatin analogue had the lowest hazard for disease progression [22]. 177Lu‐DOTATATE‐based PRRT has also been shown to improve the quality of life and significant reduction of symptoms in up to 40–70% patients. Additionally, improvement in symptoms, global health status, and Karnofsky performance status is shown to occur irrespective of the treatment outcome [23]. This is important, especially in the setting of functional tumors, where the symptoms lead to significant distress and reduction in the quality of life. The combined benefit of increased survival with symptomatic improvement leads to increased overall patient satisfaction with the therapy. Combination of PRRT with chemotherapy, such as capecitabine, or targeted therapy, such as sunitinib or everolimus, has shown to have better efficacy than monotherapies, although proof from more large‐scale studies is needed before definite recommendations can be made [24–27]. Multiple studies have shown the overall safety of 177Lu‐DOTATATE‐based PRRT. An overview of the adverse effects associated with PRRT is provided in Table 32.6. The acute side effects, such as nausea and vomiting, are related to the amino acid infusion not the radionuclide therapy, and are well managed with antiemetics. Moreover, the newer amino acid formulations are minimally emetogenic. Hormonal crises after PRRT in functional NENs is a rare adverse effect, with reported occurrence in <1% of the patients undergoing treatment. These patients are typically managed with a high dose of octreotide (500–1000 μg intravenously) along with intravenous fluids, corticosteroids, and maintenance of normal electrolyte levels. Subsequent cycles of therapy should also be given under the cover of corticosteroids and octreotide [29]. Bone marrow suppression is the most common subacute adverse effect after PRRT. The transient hematologic toxicity most commonly includes thrombocytopenia, as well as anemia and leukopenia [30]. Concurrent administration of positively charged amino acids during PRRT has significantly lowered the incidence of post‐therapy nephrotoxicity. However, despite that, creatinine clearance loss of around 4% per year has been observed in patients undergoing 177Lu‐DOTATATE‐based PRRT [27]. Table 32.6 Acute, subacute and long‐term adverse effects with 177Lu‐DOTATATE‐based PRRT Source: Based on Kwekkeboom and Krenning [27] and Bergsma et al. [28]. a MDS, myelodysplastic syndrome. The occurrence of serious adverse effects such as myelodysplastic syndrome (MDS) and acute leukemias is generally low after PRRT, although concerns of under‐reporting of these conditions due to lack of long‐term follow‐up exist [27]. Factors such as older age group (>70 years), prior treatment with alkylating agents or radiotherapy/extensive pre‐treatment, skeletal metastases, and baseline cytopenias have been shown to predispose occurrence of MDS/acute leukemias post treatment with PRRT [31, 32]. Prostate cancer (PC), the second most commonly diagnosed malignancy, forms the fifth leading cause of cancer‐related mortality in men worldwide [33]. Despite screening practices for early disease detection and availability of advanced treatment options, current challenges associated with management of PC include advanced disease at presentation, early and multifocal disease recurrence, and evolution of castrate‐resistant PC (CRPC). While a variety of molecular targets for PC are being evaluated (see future directions below), the greatest focus and currently available literature is on prostate‐specific membrane antigen (PSMA), a 750 amino‐acid transmembrane protein, overexpressed in PC cells, which will be the focus of this section. After initial attempts of targeting PSMA with antibodies [34], a breakthrough was achieved with small molecule urea‐based PSMA inhibitors (PSMA‐11) that provided superior imaging characteristics [35–38]. Additionally, multiple PSMA ligands such as PSMA‐617 and PSMA‐I&T have been labeled with 177Lu and 225Ac for PSMA‐based radioligand therapy (RLT) in PC [39–43]. In the present section, we review the current status and future directions of PSMA‐based theranostics in PC. Our focus will be on 177Lu‐PSMA RLT as per most of the literature available to date. Functional imaging targeting PSMA has been explored for multiple indications in patients with PC (Figure 32.4). Table 32.7 provides a list of indications in PC where PSMA imaging has shown promise [44, 45]. Figure 32.4 64‐year‐old male with adenocarcinoma of prostate (Gleason 5 + 4 = 9) post neo‐adjuvant hormonal therapy as well as chemotherapy underwent 68Ga‐PSMA‐11 PET/CT (maximum intensity projection, A) showing multiple PSMA expressing metastases, including nodal (iliac, B, C: arrow‐head; para‐aortic D, E: thin arrow), pulmonary, and skeletal (F, G, thick arrow). This patient would be a potential candidate for 177Lu‐PSMA‐617 radioligand therapy. Table 32.7 Present and evolving indications of PSMA‐based functional imaging 177Lu‐PSMA RLT is not currently approved by the US FDA so its use is limited to research trials. However, it is expected to be approved in the coming year(s). As per the theranostics principle described in the prior section, demonstration of adequate PSMA expression on the tumor sites is a pre‐requisite for consideration for 177Lu‐PSMA RLT and is performed by either PET‐ or SPECT‐based PSMA targeted imaging. The degree of PSMA expression on the tumor cells should be higher (ideally 1.5×) than physiologic PSMA expression, as in the liver [46]. An overall assessment of the patient has to be made, including performance status (Eastern Cooperative Oncology Group [ECOG] score < 2), life expectancy (>6 months), and symptomatology, in the context of a multidisciplinary tumor board. A baseline dynamic renal scintigraphy (99mTc‐MAG3/99mTc‐EC/99mTc‐DTPA) can be performed to rule out any underlying urinary tract obstruction. Any potentially myelosuppressive therapy should be withdrawn at least 6 weeks prior to RLT to prevent serious hematotoxicity [47]. The laboratory investigations required for eligibility for therapy are provided in Table 32.8 with the reference cutoff values. Adequate hydration must be ensured prior to administering 177Lu‐PSMA for rapid urinary excretion of the unbound tracer and reduction in the absorbed dose to kidneys. One to two liters of intravenous normal saline maybe administered at 20 mL/min in patients with low cardiovascular risk [46]. Patients with urinary incontinence or lower urinary tract symptoms due to prostatomegaly should be identified prior to therapy and duly catheterized. Pretherapy with corticosteroids is required in patients with closed/narrow cavity metastases where an obstructive swelling may develop after therapy (such as cerebral or spinal canal metastases). Several measures, such as application of cold packs, PSMA inhibitors, anticholinergics, and sialendoscopy, have been tried to prevent post‐therapy xerostomia, but no conclusive results have been obtained to recommend their routine use and these should be patient‐specific [48, 49]. Table 32.8 Laboratory investigations prior to 177Lu‐PSMA RLT Source: Based on Kratochwil et al. [46]. 177Lu‐PSMA is administered as a slow intravenous injection at a dose of 6–8.5 GBq per cycle [46]. The administration may be done either as a slow injection by hand or by an infusion pump. Diuretics and laxatives may be used to facilitate rapid excretion of the unbound radiotracer. A post‐therapy whole‐body scintigraphy (with or without SPECT) performed immediately or within 48 hours after therapy can be done to confirm radiotracer uptake at the involved sites. The blood counts should be repeated every 2–3 weeks after administration of RLT to monitor for hematotoxicity. Liver and renal function tests should be reassessed prior to the initiation of each cycle, along with physical examination and assessment of symptoms, their severity, and quality of life. Follow‐up PSA and imaging (PSMA PET/CT or PET/MRI) may be performed after every two cycles for response assessment [46]. A repeat therapy may be planned after 6–8 weeks, which allows for recovery of any affected hematologic parameters. Up to six cycles of 177Lu‐PSMA RLT are typically done without any dose‐limiting renal toxicity [50]. The efficacy of 177Lu‐PSMA RLT is based on PSA decline, objective response based on imaging, improvement in symptoms and quality of life, and effect on survival. Meta‐analyses have reported 68–75% rate of patients having any decline in PSA post 177Lu‐PSMA RLT. A >50% decline in PSA levels has been seen in 34–51% of patients [51–53]. In contrast, docetaxel‐based chemotherapy, which forms the first line of treatment in metastatic CRPC (mCRPC), has shown a >50% decline in PSA levels in ~48% of patients [54]. Cabazitaxel in the Phase 3 TROPIC trial showed >50% decline in PSA level in ~39.2% of patients. The FIRSTANA trial comparing docetaxel and cabazitaxel as first‐line therapy in mCRPC showed >50% decline in PSA levels in ~68.4% patients treated with docetaxel versus ~68.7% in the cabazitaxel group. However, patients currently undergoing 177Lu‐PSMA RLT belong to a heavily pretreated population, having exhausted standard of care therapies, and thus a direct comparison of these parameters should be made with caution. Pooled results from eight studies on 175 patients have shown PR in 37.2%, stable disease (SD) in 38.3%, and progressive disease (PD) in 24.5% of patients [52]. In comparison, PR was noted in 40.5%, SD in 42.8%, and PD in 2.3% of patients receiving cabazitaxel as first‐line therapy in mCRPC [55]. The median overall survival with 177Lu‐PSMA RLT is reported at ~8.3 months in patients with mCRPC, compared to ~18.7 months with docetaxel therapy [52, 54]. However, again, considerable heterogeneity exists in the patient population of the two groups with regards to the temporal stage of the disease and prior treatment. 177Lu‐PSMA RLT leads to significant symptomatic improvement in patients, with pain due to osseous metastases at baseline. A Phase 2, single‐arm trial showed that 177Lu‐PSMA RLT led to improvement in pain in all time points in all patients with pain at baseline. The improvement in pain was both in terms of severity and the duration. Additionally, improvement was noted in cognitive functioning and insomnia during the treatment course [56]. The phase 3 prospective multicentre randomized registry trial of 177Lu‐PSMA‐617 (Endocyte’s VISION trial) has concluded and the results are due shortly. The early results are quite promising and are likely to result in FDA approval in the coming year(s). Table 32.9 Adverse effects with 177Lu‐PSMA radioligand therapy in prostate cancer Overall, 177Lu‐PSMA RLT has been shown to be an effective therapy in terms of achieving symptomatic improvement as well as a reduction in tumor burden in patients with mCRPC. 177Lu‐PSMA‐based RLT has been primarily explored in heavily pretreated patients with mCRPC. These patients are already exposed to multiple adverse effects resulting from chemotherapeutic drugs, such as docetaxel and cabazitaxel, including but not limited to hematologic toxicity, cardiotoxicity, paraesthesias, alopecia, and GI toxicity [54]. In contrast, 177Lu‐PSMA RLT is well tolerated with fewer side effects and a low incidence of grade 3–4 toxicities [56, 57]. Xerostomia is a commonly reported adverse effect, resulting from physiologic PSMA expression in the salivary glands. However, most patients experience grade I xerostomia, which is not dose limiting. The incidence of grade 3 hematotoxicity and nephrotoxicity is low, and grade 4 toxicities are extremely rare [58–60]. Table 32.9 provides a list of commonly associated adverse effects with 177Lu‐PSMA RLT. meta‐Iodobenzylguanidine (mIBG), a guanethidine analogue of norepinephrine, targets the norepinephrine transporter (NET) which is required for the reuptake of norepinephrine at the presynaptic terminals [63]. This reuptake process is primarily driven by a saturable, active adenosine triphosphate‐dependent system, and to a lesser degree by a passive diffusion pathway [64]. Once inside the tumor cells, mIBG is stored in neurosecretory granules (pheochromocytoma, paraganglioma [PPGL]) via the vesicular mono‐amine transporter or stored in the cytoplasm/mitochondria (neuroblastoma). These tumors derived from the primordial neural crest (e.g. neuroblastoma) or sympathetic nervous system (e.g. PPGLs) show increased expression of NET and thus are potential targets for mIBG‐based imaging and therapy. Neuroblastoma is the most common extracranial solid malignancy of childhood, with most cases occurring in infants and children [65]. The majority of cases present with an abdominal tumor, with around 35% having regional nodal involvement at diagnsosis [66]. Distant metastases are present in 60–70% of patients, requiring aggressive, multimodality treatment (chemoradiation, autologous stem cell transplant). Disease staging is one of the important prognostic factors in neuroblastoma, affecting treatment and survival outcomes [67]. 123I/131I‐mIBG‐based imaging is used for the detection of distant metastases prior to considering the patient for surgery and is the gold standard for staging and follow‐up of neuroblastoma patients [67, 68]. In patients with high‐risk metastatic disease, mIBG‐based imaging is also a pre‐requisite for evaluation toward high‐dose 131I‐mIBG therapy. PPGLs originate from the chromaffin cells in the adrenal medulla or the extra‐adrenal sympathetic and parasympathetic paraganglia [69]. The functional tumors, accounting for over 50% of all PPGLs, present with adrenergic symptoms (hypertension, deadache, palpitations, arrhythmias), which contribute to significant morbidity and mortality [70, 71]. Surgery is the first‐line and potentially curative treatment for localized, primary PPGLs. However, around 10% of patients have unresectable primary tumor at diagnosis, while ~35% present with distant metastases, bearing poor prognoses [72]. These patients are treated with cyclophosphamide‐, vincristine‐, and dacarbazine‐based chemotherapies, which have limited efficacy in terms of objective tumor response rates (complete response [CR] ~4%, PR ~37%) [73]. 131I‐mIBG therapy is of value in these patients, having the potential of reducing the morbidity and mortality, and achieving sustained objective tumor response. 123I‐mIBG is preferred over 131I‐mIBG for diagnostic imaging because of its favorable emissions (159 vs. 364 keV) and shorter half‐life (13.2 hours vs. 8 days), allowing earlier imaging with a lower radiation burden and permitting higher injectable doses, thereby improving sensitivity. Addition of SPECT/CT to planar imaging improves diagnostic accuracy and anatomic localization of the tumors. 131I‐mIBG imaging is then primarily reserved for pretherapy dosimetry. Oncologic indications of 123I‐mIBG imaging are provided in Table 32.10 (see also Figure 32.5). It should be noted that SSTR PET has an evolving role for PPGLs since many of these tumors, especially those with succinate dehydrogenase complex (SDHx) gene mutations, express SSTRs and given the higher resolution of PET over SPECT. However, SSTR PET would not, of course, inform about eligibility for mIBG therapy so it can only be used for diagnostic imaging purposes. Table 32.10 Oncologic indications of 123I‐mIBG imaging Source: Based on Bombardieri et al. [74]. 131I‐mIBG therapy is indicated in inoperable or metastatic tumors with sufficient mIBG avidity, as documented on pretherapy 123I/131I‐mIBG imaging. Baseline results from conventional imaging (ultrasound, CT, MRI) and biochemical tumor markers should be available for response assessment post‐therapy. The primary objectives of 131I‐mIBG therapy vary per patient, but commonly include complete or partial disease remission, preventing tumor progression, and relief of functional symptoms, including the reduction of hypertensive medications. It is important to be aware of two different 131I‐mIBG preparations: low and high specific activity (LSA and HSA). HSA 131I‐mIBG (specific activity ~92.5 MBq/μg) should be preferred wherever available over LSA (specific activity ~1.59 MBq/μg) as HSA has higher efficacy (increased radiation dose delivered to the tumor per unit injected activity) and lower incidence of cardiovascular adverse effects (low levels of unlabeled mIBG) [75]. The laboratory investigations required prior to therapy are provided in Table 32.11 with the reference values. Figure 32.5 Eight‐year‐old female with metastatic left retroperitoneal neuroblastoma treated with multiple therapies, including resection, chemotherapy, external beam radiation, stem cell transplant, and immunotherapy, presented with a left posterior skull mass concerning for recurrent disease. Planar whole‐body 123I‐mIBG scintigraphy images in anterior (A) and posterior (B) projections show widespread osseous metastases with prominent tracer uptake in the occiput (thick arrow) and right pelvis (thin arrow). She was treated with 131I‐mIBG therapy and follow‐up anterior (C) and posterior (D) 123I‐mIBG scans obtained 1‐month after therapy show a marked improvement. Table 32.11 Laboratory investigations prior to 131I‐mIBG therapy Source: Based on Gonias et al. [76], Pryma et al. [77], and Giammarile et al. [78]. Therapeutic dose of 131I‐mIBG varies across institutions. A weight‐based activity of 450–1850 MBq/kg or a standard activity of 7 GBq is used in children with neuroblastoma, with the availability of autologous hematopoietic stem cell rescue (AHSCR) for myelosuppression [79]. Adult patients undergoing treatment for PPGLs typically receive 400–500 mCi (high‐dose regimens >600 mCi with AHSCR) 131I‐mIBG per treatment cycle [77, 79]. Bone marrow is the dose‐limiting organ in 131I‐mIBG therapy, thus the injected activity requires reduction in patients with myelosuppression or impaired renal function and is based on patient‐specific pretherapy dosimetry using serial whole‐body scintigraphy. Higher activities may be injected when AHSCR support is available. The vital signs (blood pressure [BP], pulse rate) of the patient must be measured immediately prior to, during, and at frequent intervals after therapy. This is especially important for PPGLs, where mIBG administration can lead to unstable BP. Sudden elevation of BP during therapy can be commonly managed by temporarily halting the infusion. Alpha‐ or beta‐blockers may be required in refractory cases. 131I‐mIBG diluted in normal saline is infused slowly (over 45 minutes to 4 hours) via an indwelling intravenous cannula under a lead‐shielded infusion system. The infusion line is flushed with normal saline after completion of 131I‐mIBG infusion at the same initial rate [78]. Post‐therapy whole‐body scintigraphy (with or without SPECT/CT) is typically done to confirm tracer uptake in the lesions (delayed imaging at/ after ~4 days) and can also be done for post‐therapy dosimetry (serial imaging) [78, 79]. The patients must be monitored regularly for hematologic toxicities, especially during the first 6 weeks after therapy (weekly or higher frequency, depending on administered dose). Temporary myelosuppression is expected in the majority of the patients, but frequent monitoring can help in early institution of supportive therapies such as granulocyte‐colony stimulating factors (G‐CSF), platelets, whole‐blood transfusion, and erythropoietin. Patients receiving high‐dose therapy are more likely to develop prolonged myelosuppression and require AHSCR, the harvesting of which has to be planned and executed prior to therapy [76]. Response evaluation using conventional imaging (CT, MRI) and biochemical tumor markers is typically performed at 3–6 months after treatment. 123I‐mIBG imaging can also be done to assess functional response. Retreatment can be planned as early as 3–6 months, provided the blood parameters have recovered and meet the baseline requirement for therapy [80, 81]. 131I‐mIBG therapy is instituted mostly in pretreated patients with stage III–IV neuroblastoma, where the prognosis is already poor and the patients are at high‐risk of developing adverse effects such as myelosuppression. In a phase II study of 163 patients treated with relapsed/ refractory neuroblastoma, treated with LSA 131I‐mIBG, 8% had CR, 28% had PR (overall objective response rate 36%) and 27% had PD. The overall survival at 1 and 2 years was 49% and 29%, respectively [82]. A phase II study using 131I‐mIBG therapy in combination with topotecan in 16 treatment‐naïve patients with high‐grade neuroblastoma showed an overall objective response rate of 57% and primary tumor response rate of 94% after two treatment courses [83]. A systematic review of 131I‐mIBG therapy in relapsed/refractory neuroblastoma showed an overall mean tumor response rate of 32% with an overall survival at 1 year ranging from 38% to 100% [84]. The currently available data suggests that high‐dose 131I‐mIBG therapy (18 mCi/kg body weight) combined with AHSCR can improve survival and objective tumor response rates in children with grade III/IV neuroblastoma [85, 86]. A phase II study of LSA 131I‐mIBG in 50 patients with metastatic PPGLs showed 8% CR and 14% PR. An overall response rate, including patients with some degree of tumor regression was 57% [76]. HSA 131I‐mIBG therapy has been explored in unresectable/metastatic/recurrent PPGLs. A phase II clinical trial showed that 92.2% of patients had PR or stable disease as the best tumor response, with no complete responses. Twenty‐five percent of patients could achieve BP control, with the majority receiving two treatment doses [77]. These results were superior to those observed from cyclophosphamide‐, vincristine‐, and dacarbazine‐based chemotherapy in metastatic PPGLs (4% CR, 37% PR) [73]. The results of this study paved the way for FDA approval of HSA 131I‐mIBG in 2018 for metastatic mIBG‐avid PPGLs. Myelosuppression is the most concerning toxicity associated with 131I‐mIBG therapy. Early, temporary myelosuppression is typically observed at 2–4 weeks of therapy, with the nadir values occurring at 4–6 weeks [79]. G‐CSF support, whole‐blood or platelet transfusions may be required to manage early grade 3–5 toxicities. Around 6–33% of patients may require AHSCR due to persistent myelosuppression [76, 82, 87]. The late adverse effects are similar to those observed with radioiodine (131I‐NaI) therapy for thyroid cancer. Despite adequate thyroid blockade, abnormalities in thyroid function tests are observed in up to 22% of patients undergoing 131I‐mIBG therapy [88]. Several secondary hematologic and solid tumors have been described in patients on long‐term follow‐up after 131I‐mIBG therapy. However, establishing a definite causality is difficult in these heavily pretreated patients [79–90]. Table 32.12 provides a list of the early and late adverse effects reported with 131I‐mIBG therapy. Table 32.12 Early and late adverse effects with 131I‐mIBG therapy Values in parentheses represent incidence rates. a Other toxicities: acute respiratory distress syndrome, bronchiolitis obliterans organizing pneumonia, pulmonary embolism, hypogonadism, GI adverse effects, hypotension, hepatotoxicity, infections, fever. Pain resulting from osseous metastases is frequently debilitating and a major source of morbidity in patients with various malignancies. The pathophysiologic basis of metastatic osseous pain in the absence of a fracture is multifactorial and likely the result of an interplay of several factors such as periosteal dilatation, nerve entrapment/compression, mechanical stress on the bone, and release of cytokines and other algesic chemicals in the marrow [95]. This partly explains why the occurrence and severity of pain is not correlated with the size and number of metastases, type of primary tumor or its location [96]. The palliation of osseous pain involves multimodality management involving pharmacologic anti‐inflammatory and analgesic agents, local and systemic anticancer medical therapies, invasive procedures, radiation therapy, and radionuclide therapies [97]. Pharmacologic therapy, including nonsteroidal anti‐inflammatory drugs and various opioids, is typically the initial treatment of choice. Surgery and locally directed therapies are useful in cases of solitary painful metastasis or limited oligo‐metastatic disease. Radiation therapy provides immediate relief and is useful in the setting of osseous pain with imminent risk of fracture or spinal cord/neural compression. The adverse effects with radiation therapy, especially with total body or hemi‐body irradiation, include severe myelo‐suppression, and GI and local toxic effects [95]. However, for disseminated skeletal metastases, systemic therapies such as palliative chemotherapy or radionuclide therapy are best suited. Radionuclide therapy has the advantage of lower incidence of adverse effects, ease of therapy administration, prolonged pain relief, and the ability to target all (including subclinical) sites of osseous metastases [95, 98
32
Radionuclide Therapies and Correlative Imaging
Introduction
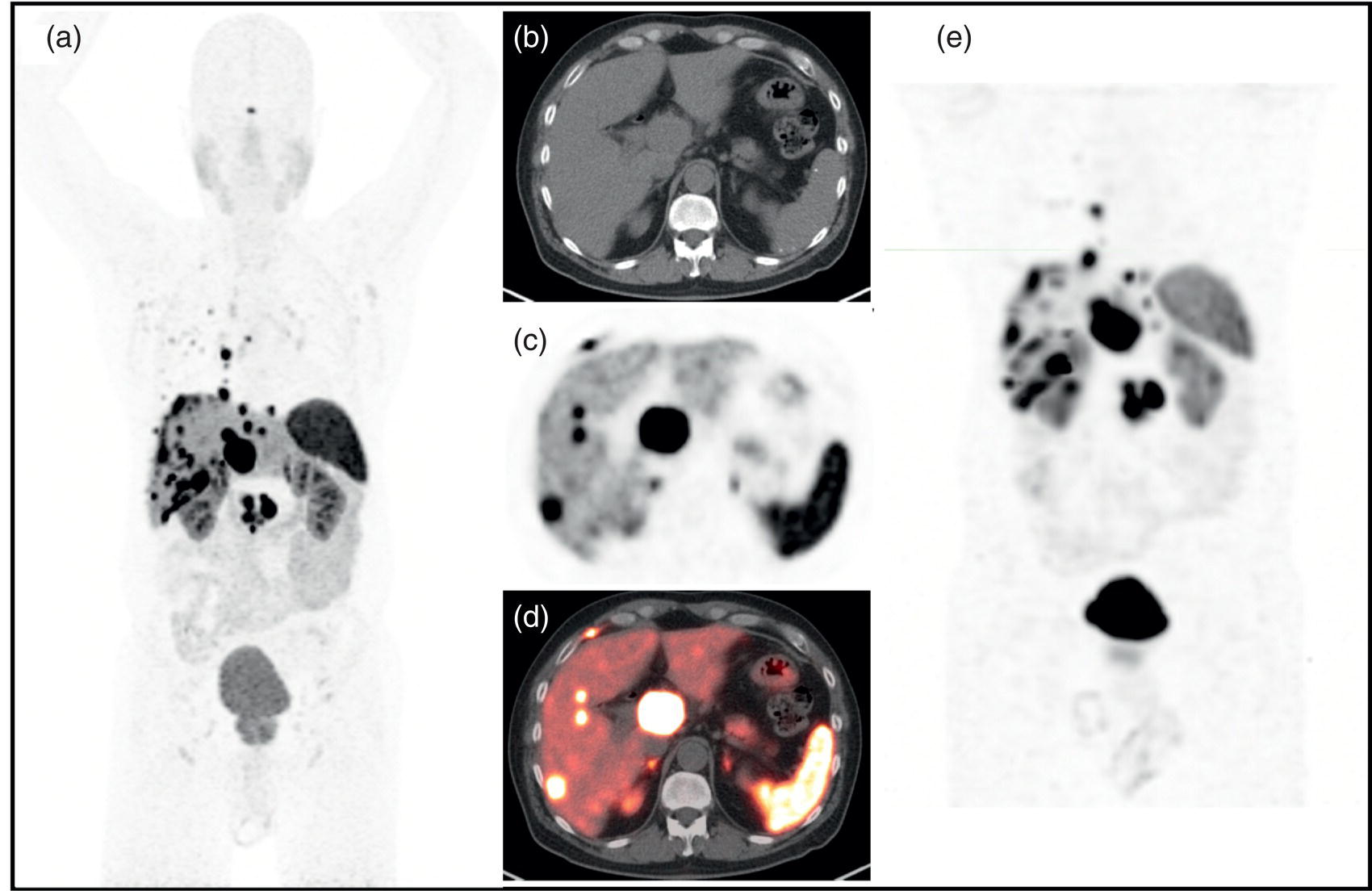
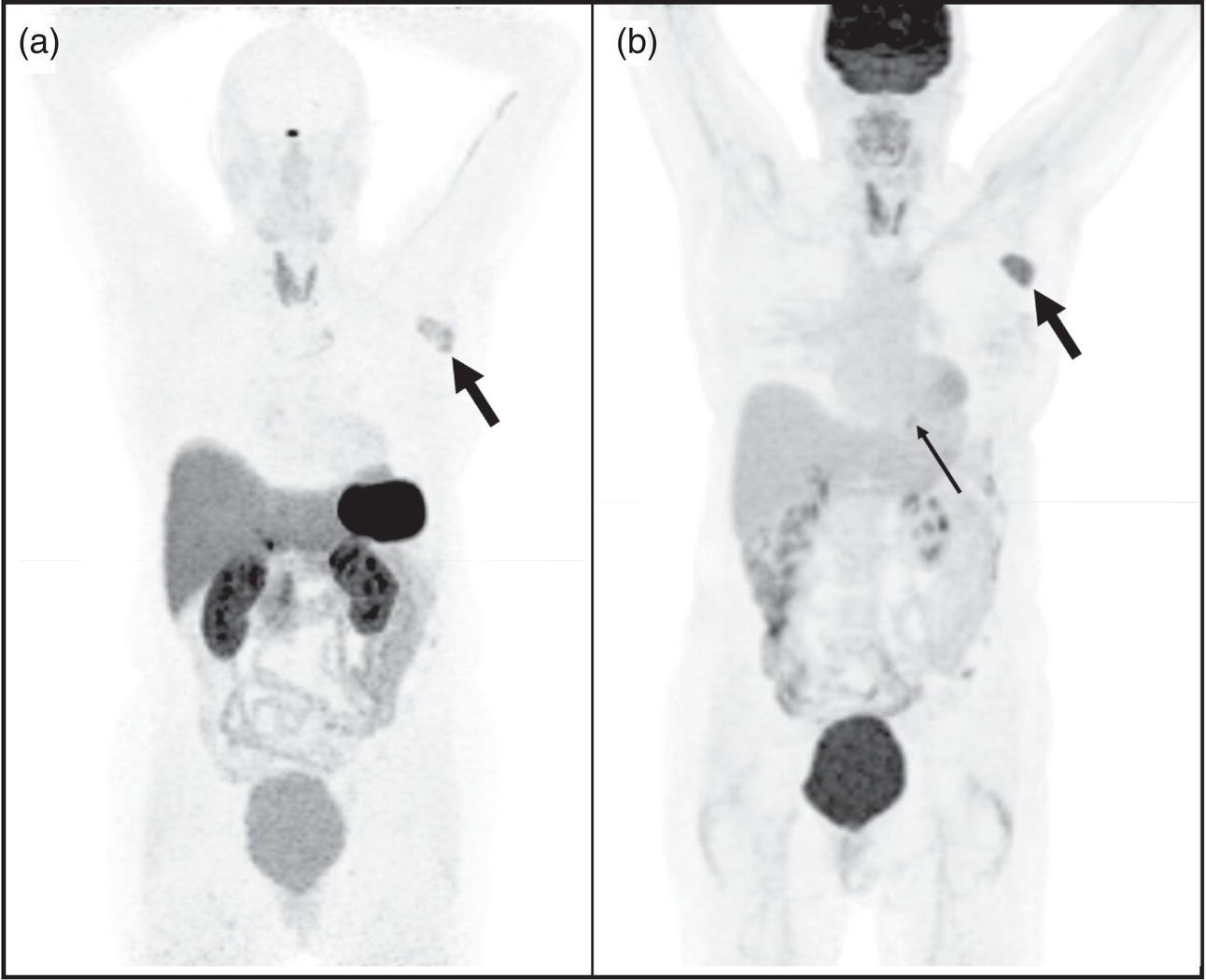
Theranostics in Neuroendocrine Neoplasms
Introduction
Objectives of Functional Imaging

Clinical setting
Indication
Suspected NEN
Clinical or biochemical suspicion of NEN with inconclusive findings on conventional imaging and nonavailability of a histologic diagnosis
Raising the diagnostic confidence in lesions not amenable to biopsy, but otherwise showing typical characteristics of NEN on conventional imaging
Selection of biopsy site in patients with suspected NEN and previous inconclusive conventional image‐guided biopsy
Histologically confirmed NEN, baseline evaluation
Initial staging of histopathologically confirmed NEN
Detection of primary tumor in patients with confirmed metastasis
Assessment for suitability of PRRT in surgically unfit candidates
Histologically confirmed NEN, re‐evaluation
Assessment of residual disease after surgery
Restaging in patients with clinical or biochemical suspicion of disease recurrence
Assessment of new lesions in previously treated patients with NEN and inconclusive findings
Assessment for suitability of PRRT as an adjuvant therapy
Assessment of response to PRRT
Radionuclide Therapy: Indications and Procedure
Clinical setting
Goal of therapy
Neo‐adjuvant
To achieve surgical eligibility in presently inoperable NENs
Adjuvant
Disease control in cases with incomplete resection of the primary NEN
Disease control at the time of recurrence after initial surgery
Unresectable, Metastatic NEN
Disease control
Symptom control in functional tumors
Salvage PRRT
Disease control in patients previously treated with PRRT, presenting with recurrence
Tumor grade
Mitotic activity (per 2 mm2)a
Ki‐67 labeling index
G1 (low)
<2
<3%
G2 (intermediate)
2–20
3–20%
G3 (high)
>20
>20%
Laboratory test
Acceptable value at baseline
Hemoglobin
>8 g/dL
White blood count
>2000/mm3
Platelet count
>70 000/mm3
Total bilirubin
<3 times the upper limit of normal
Albumin
>3 g/dL
Estimated glomerular filtration rate (eGFR)
>50 mL/min
Pretreatment
Therapy Administration
Parameter
Requirement
Lysine content
18–24 g
Arginine content
18–24 g
Volume
1.5–2.2 L if commercial (1 L if compounded)
Osmolarity
<1050 mOsmol
Post‐therapy: Discharge and Follow‐Up
Efficacy
Safety
Acute
Subacute
Long‐term
Nausea (25%)
Grade 3 or 4 hematologic toxicity (<15%)
Renal insufficiency (<1%)
Vomiting (10%)
Temporary hair loss (60%)
MDSa (1–4%)
Abdominal pain (10%)
Hormonal crisis (<1%)
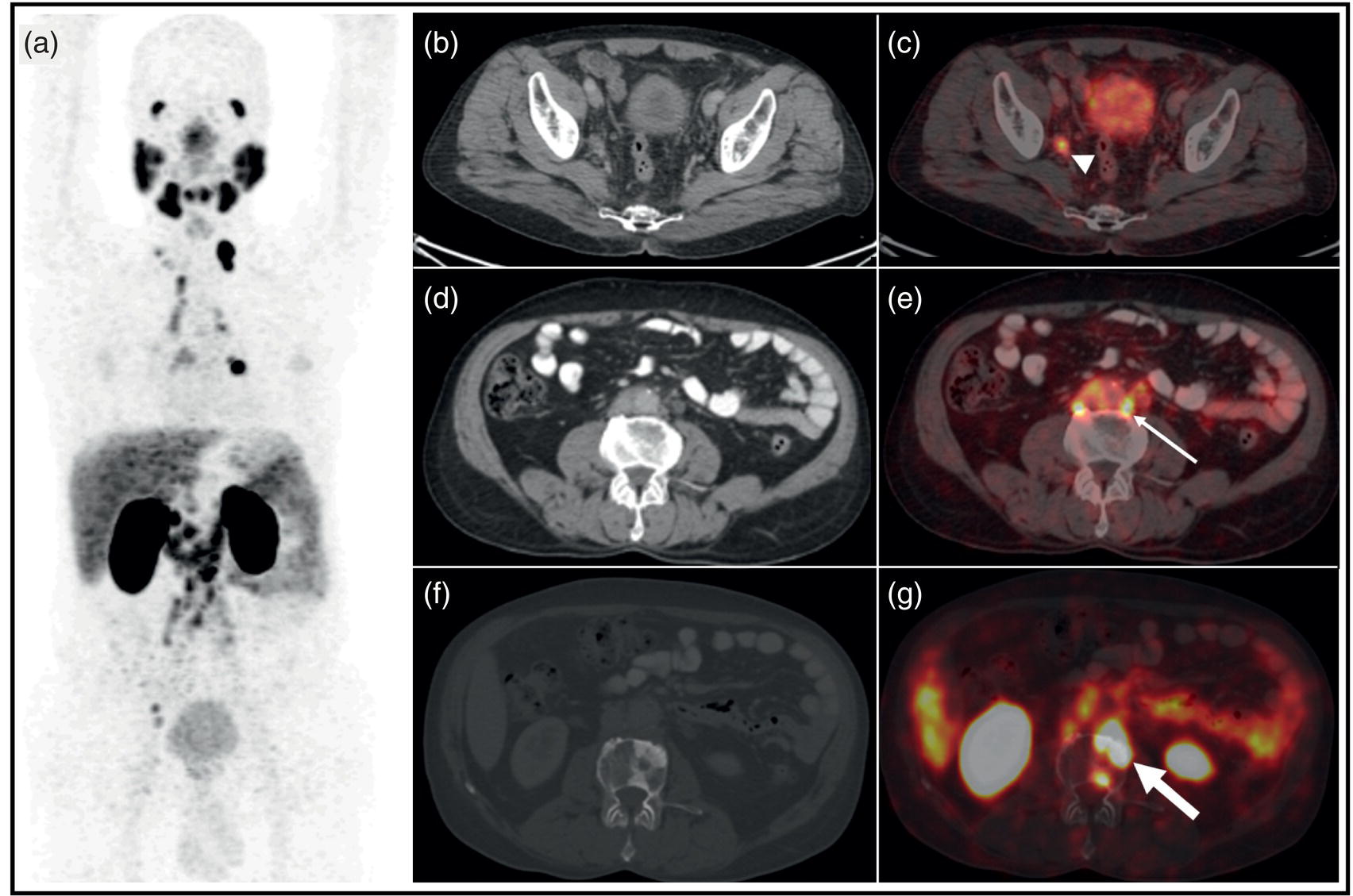
Theranostics in Prostate Cancer
Introduction
Objectives of Functional Imaging
Clinical setting
Indication
Suspected PC
High clinical or biochemical suspicion of PC with inconclusive findings on conventional imaging and negative or unavailable histologic diagnosis
Site selection for image‐guided biopsy in patients with suspected PC and previous inconclusive biopsy
Histologically confirmed PC, baseline evaluation
Initial staging of histopathologic confirmed, high‐risk PC (T3a, or Gleason score ≥ 7, or PSA > 20 ng/mL)
Histologically confirmed PC, re‐evaluation
Biochemical recurrence (detection of the site of recurrence and identifying potential candidates for salvage therapy)
Assessment of suitability for PSMA RLT
Response assessment after PSMA RLT
Radionuclide Therapy: Indications and Procedure
Pre‐treatment
Laboratory test
Acceptable value at baseline
White blood count
>2500/mm3
Platelet count
>75 000/mm3
Liver transaminases
<5 times the upper limit of normal
Serum creatinine
<2 times the upper limit of normal
Estimated glomerular filtration rate (eGFR)
>30 mL/min
Therapy Administration
Post‐therapy: Discharge and Follow‐Up
Efficacy
Adverse effects
Incidence
Salivary gland toxicity [56, 61]
0–87%
Hematologic toxicity [57]
Leukopenia
All grades ~40%, grade 3–4 ~3%
Anemia
All grades ~34%, grade 3–4 ~10%
Thrombocytopenia
All grades ~31%, grade 3–4 ~4%
Nephrotoxicity [61, 62]
All grades 4.5–93%
Safety
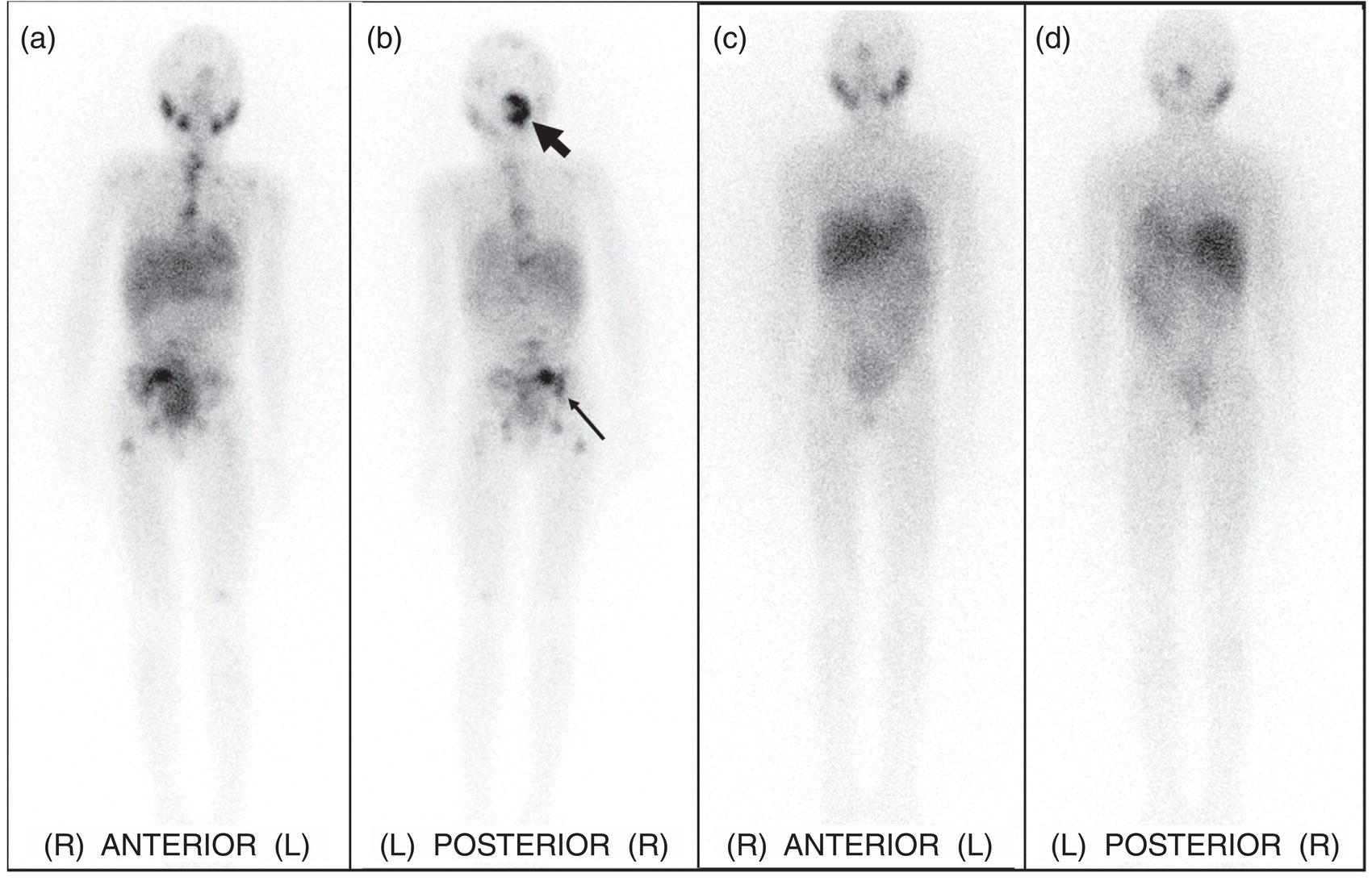
mIBG‐based Theranostics in Neuroblastoma and Pheochromocytoma/Paraganglioma
Introduction
Objectives of Functional Imaging
Clinical setting
Indication
Suspected neuro‐endocrine origin tumors
Imaging‐based confirmation of mIBG avid tumors: neuroblastoma, ganglioneuroblastoma, ganglioneuroma, PPGL, carcinoid, Merkel cell tumors, and medullary thyroid cancer
Confirmed NENs
Initial staging and restaging
Unresectable/metastatic tumors
Evaluation for high‐dose 131I‐mIBG‐based therapy: eligibility and dosimetry
Evaluation of response to therapy
Radionuclide Therapy: Indications and Procedure
Pretreatment
Laboratory test
Acceptable value at baseline
White blood count
>3000/mm3
Absolute neutrophil count
≥1000/mm3
Platelet count
>80 000/mm3
Liver transaminases
≤2.5 times the upper limit of normal
Serum creatinine
≤2.5 times the upper limit of normal
Estimated glomerular filtration rate (eGFR)
>30 mL/min
Therapy Administration
Post‐therapy: Discharge and Follow‐Up
Efficacy
Safety
Early
Late
Nausea, vomiting (~21–78%) [77, 91]
Hypothyroidism (~5% requiring thyroxine supplementation) [88]
Temporary myelosuppression (~60–87%) [76, 78, 92]
Persistent myelosuppression (~6–33%) [76, 82]
Transient sialadenitis (~41–90%) [77, 93]
Secondary hematologic/solid tumors (~4–14%) [79, 87, 89, 90]
Hypertensive crisis (~0–15%) [76, 77, 94]
Othersa [76, 77, 87, 91]
Radiopharmaceutical Therapies for Pain Palliation from Osseous Metastases
Introduction
Related posts:
 Nephro‐urinary Tract Pathologies: A Correlative Imaging Approach
Nephro‐urinary Tract Pathologies: A Correlative Imaging Approach
 Lymphoma and Myeloma Correlative Imaging
Lymphoma and Myeloma Correlative Imaging
 Artificial Intelligence in Diagnostic Imaging
Artificial Intelligence in Diagnostic Imaging
 Osteoporosis: Diagnostic Imaging and Value of Multimodality Approach in Differentiating Benign Versus Pathologic Compression Fractures
Osteoporosis: Diagnostic Imaging and Value of Multimodality Approach in Differentiating Benign Versus Pathologic Compression Fractures
 The Role of Noninvasive Cardiac Imaging in the Management of Diseases of the Cardiovascular System
The Role of Noninvasive Cardiac Imaging in the Management of Diseases of the Cardiovascular System
 Spine Disorders: Correlative Imaging Approach
Spine Disorders: Correlative Imaging Approach
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


