PULMONARY ALVEOLAR PROTEINOSIS
Pulmonary alveolar proteinosis (PAP) is a rare disorder characterized pathologically by the accumulation of lipoprotein within alveoli.
There are three different forms of the disease. Congenital PAP results from mutations in genes encoding surfactant B or C or granulocyte–macrophage colony-stimulating factor (GM-CSF). Secondary PAP occurs in association with a wide variety of disorders, including acute silicosis (silicoproteinosis), infections, and hematologic and lymphatic malignancy. Idiopathic PAP accounts for nearly 90% of cases. It is an autoimmune disease in which antibodies to GM-CSF interfere with the degradation or clearance of surfactant.
Men with PAP outnumber women. Patients range in age from a few months to more than 70 years, with two-thirds of patients being between 30 and 50 years old. Symptoms are usually mild and of insidious onset. They include cough, fever, and mild dyspnea. About 30% of patients are asymptomatic.
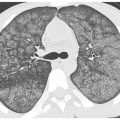
HRCT abnormalities are typically diffuse, bilateral, and often symmetric (Table 18.1). The characteristic HRCT abnormality consists of geographic regions of ground glass opacity and smooth interlobular septal thickening in the same lung regions. This is termed the crazy paving pattern (Figs. 18.1 and 18.2). Consolidation or ill-defined nodules may also be present, but are not particularly common. The differential diagnosis of the crazy paving pattern in patients with chronic symptoms is long and includes interstitial pneumonias, organizing pneumonia, eosinophilic pneumonia, hypersensitivity pneumonitis, lipoid pneumonia, and invasive mucinous adenocarcinoma.
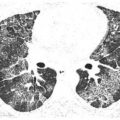
HRCT may be used to monitor treatment (Fig. 18.2). Large volume bronchoalveolar lavage is used for treatment in an attempt to clear the proteinaceous material from the alveolar spaces. HRCT can help target appropriate regions of lung for bronchoalveolar lavage and to confirm successful treatment. With lavage, ground glass opacities improve or resolve. Septal thickening may sometimes persist (Fig. 18.2B).
LIPOID PNEUMONIA
Lipoid pneumonia reflects the accumulation of fats within the lung parenchyma. It may be endogenous or exogenous. Endogenous lipoid pneumonia is characterized by lipid-laden macrophages within consolidated lung distal to an obstructed airway. HRCT findings are those of a post-obstructive pneumonia.
Exogenous lipoid pneumonia is caused by the aspiration of fatty materials such as mineral oil. As a large amount of lipid needs to be aspirated over a long period of time, patients usually present with chronic symptomatology. Also, CT abnormalities may be incidentally discovered in asymptomatic patients.
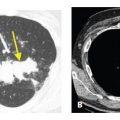
The HRCT findings of exogenous lipoid pneumonia include ground glass opacity or consolidation that is patchy, focal, or mass-like (Table 18.2). A combination of ground glass opacity and interlobular septal thickening, the crazy paving pattern, is not uncommon. Soft tissue windows may show low-attenuation regions within areas of consolidation, with Hounsfield unit measurements consistent with fat (Fig. 18.3). Masses or regions of consolidation may be round or irregular in shape.
Three forms: congenital, secondary, idiopathic (90% of cases)
Diffuse and bilateral
Patchy and geographic regions of GGO and crazy paving
Consolidation less common
GGO decreases with bronchoalveolar lavage
GGO, ground glass opacity.
Findings are often most severe in the dependent lung regions, reflecting the typical distribution of aspiration. Associated fibrosis may be present, although this is usually not a significant component of disease.
AMYLOIDOSIS
Amyloidosis reflects the accumulation of an extracellular protein in multiple organs. The most commonly involved organs include the kidneys, heart, nervous system, and liver.
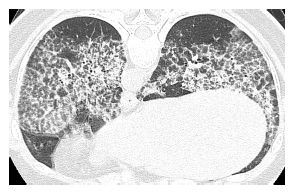
Figure 18.1
Pulmonary alveolar proteinosis. Prone HRCT shows a combination of ground glass opacity and interlobular septal thickening in the same lung regions, the crazy paving sign. In a patient with chronic symptoms, alveolar proteinosis is one of several diagnoses that can produce this pattern.
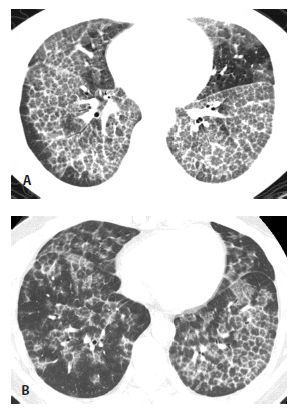
Figure 18.2
Pulmonary alveolar proteinosis, before and after treatment. A. Typical findings of alveolar proteinosis with crazy paving are present on pretreatment HRCT. B. After several treatments with large volume bronchoalveolar lavage, there has been significant improvement in the ground glass opacity and septal thickening.
There are two types of amyloid that may be deposited in the lungs, amyloid light chain and amyloid A chain. The former is responsible for the majority of patients with symptomatic lung involvement; it may be idiopathic or associated with myeloma or lymphoma. A chain amyloidosis is associated with rheumatoid arthritis, inflammatory bowel disease, chronic inflammatory disorders (e.g., osteomyelitis), and chronic pulmonary infections (e.g., tuberculosis) and rarely leads to symptoms.
Table 18.2 HRCT findings of lipoid pneumonia
Aspiration of fatty materials (exogenous lipoid pneumonia)
Patchy and dependent
Ground glass opacity or consolidation
Low-attenuation consolidation
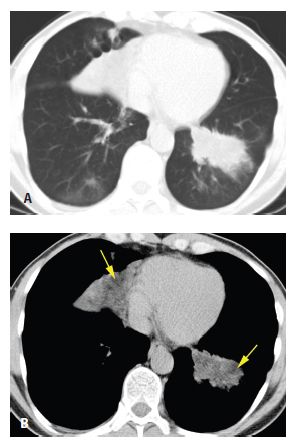
Figure 18.3
Lipoid pneumonia. A. Lung windows show focal regions of consolidation at both lung bases in a patient with chronic symptoms. This finding is nonspecific. B. The presence of low-attenuation fat (arrows) within regions of consolidation on the mediastinal windows strongly suggests the diagnosis of lipoid pneumonia.
Table 18.3 HRCT findings of amyloidosis
Focal parenchymal amyloidosis
Single or multiple lung nodules, sometimes calcified
Diffuse parenchymal amyloidosis
Small nodules with a perilymphatic distribution, septal thickening
Consolidation, ground glass opacity, calcification, fibrosis
Tracheobronchial amyloidosis
Diffuse or focal thickening tracheal and bronchial walls
Calcification common
Amyloidosis may involve the lung parenchyma or airways (Table 18.3). Lung parenchymal involvement may be focal or diffuse. Lymphadenopathy may be associated with any of the above patterns of amyloid or as an isolated abnormality. Calcification is often present in mediastinal or hilar lymph nodes.
Focal Parenchymal Amyloidosis
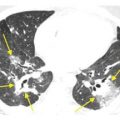
Focal parenchymal amyloidosis presents with a single or multiple discrete lung nodules or masses (Fig. 18.4). These may be calcified. They are difficult to differentiate from other causes of nodules such as primary bronchogenic carcinoma, metastases, granulomatous disease, or hamartoma. Associated airway wall thickening may be present in patients with focal parenchymal amyloid.
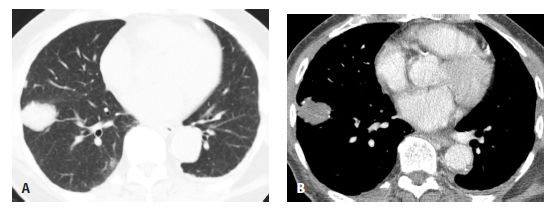
Figure 18.4
Amyloidosis, focal parenchymal. A. A nonspecific nodule is seen in the right lower lobe. B. On a soft tissue window, focal areas of calcification are visible within the nodule. Biopsy confirmed amyloidosis in this patient with rheumatoid arthritis.
Related posts:
 HRCT Indications, Technique, Radiation Dose, and Normal Anatomy
HRCT Indications, Technique, Radiation Dose, and Normal Anatomy
 Pulmonary Edema, Diffuse Alveolar Damage, the Acute Respiratory Distress Syndrome, and Pulmonary Hemorrhage
Pulmonary Edema, Diffuse Alveolar Damage, the Acute Respiratory Distress Syndrome, and Pulmonary Hemorrhage
 Hypersensitivity Pneumonitis and Eosinophilic Lung Disease
Hypersensitivity Pneumonitis and Eosinophilic Lung Disease
 Complications of Medical Treatment:Drug-Induced Lung Disease and Radiation
Complications of Medical Treatment:Drug-Induced Lung Disease and Radiation
 Reticular Opacities
Reticular Opacities
 Nodular Lung Disease
Nodular Lung Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


