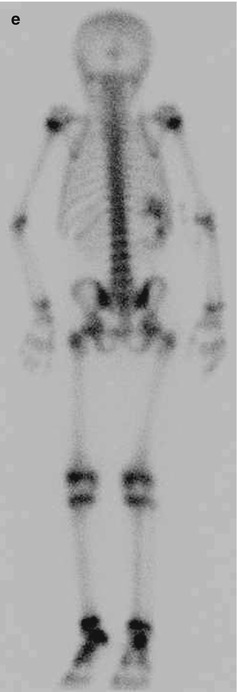
Fig. 11.1
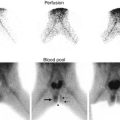
Wilms’ tumor. Grayscale ultrasound of the right kidney (a). Large heterogeneous echogenic soft tissue mass arising from right upper pole, with small cystic regions representing necrosis. Surrounding hypoechoic edge representing pseudocapsule. Color ultrasound of the IVC (b). Focal expansion of infrahepatic IVC by large heterogeneous filling defect representing neoplastic vascular invasion. Flow seen cranial to tumor thrombus. CT Abdomen, axial corticomedullary phase (c and d) Large heterogeneously enhancing upper-pole mass in the right kidney, with distortion of the right renal artery. Soft tissue expansion of the transhepatic IVC with tumor. Tc99m MDP bone scintigraphy (e). Abnormal, asymmetric renal uptake in right kidney
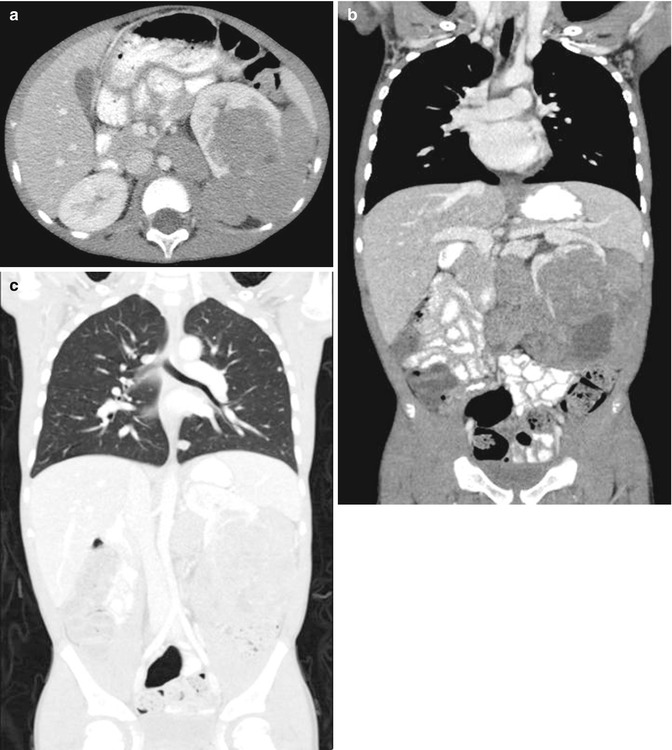
Fig. 11.2
Wilms’ tumor, blastemal cell type. CT abdomen and pelvis, axial (a) and coronal (b) nephrographic phase. Multilobular homogeneously enhancing soft tissue mass arising from the left kidney, with claw sign and distortion of the renal hilum. Retroperitoneal lymph adenopathy in the left periaortic region. Renal vein and IVC patent. CT abdomen and pelvis, coronal in lung window. (c) Several small peripheral pulmonary nodules consistent with metastases
Preoperative imaging plays a vital role in the management of Wilms’ tumor. The NWTS protocol advocates imaging prior to radical nephrectomy and adjuvant therapy, while SIOP advocates neoadjuvant chemotherapy based on initial imaging [7]. Recent reports indicate a difficulty in the accurate clinical staging of localized Wilms’ tumor when compared to pathologic results [7].
Ultrasound is usually the first imaging modality in the diagnosis of intra-abdominal masses given its safety profile. US may demonstrate an echogenic mass that is partially surrounded by a hypoechoic rim representing the pseudocapsule of Wilms’ tumor which is composed of normal compressed renal parenchyma [8]. US may demonstrate a tumor mass of mixed echogenicity with extensive surrounding necrosis. Calcification appears as areas of hyperechogenicity with shadowing [8]. IVC involvement and regional adenopathy may be detected by US although it is less sensitive than CT [8].
CT is the primary imaging modality for staging. On nonenhanced CT imaging, Wilms’ tumor appears as a large heterogeneous mass, less dense than the surrounding normal kidney [5]. Low-density areas correspond to areas of necrosis and small areas of intrarenal calcification may be identified. After administration of intravenous contrast, Wilms’ tumors show slight enhancement, a pseudocapsule that is usually sharply defined, and areas of necrosis that fail to demonstrate significant uptake of contrast [5]. In infiltrative cases of Wilms’ tumor, CT demonstrates an ill-defined mass that completely replaces the kidney with almost no normal parenchyma identified. CT scan may identify regional adenopathy; however, it cannot reliably differentiate reactive from malignant changes. Tumor staging by CT is also likely to overestimate intrarenal disease (stage I) for local extrarenal extension (stage II) [7].
On MRI, Wilms’ tumor appears as a large, solid renal tumor. The tumor appears heterogeneous with intermediate signal intensity on T1-weighted images and high signal intensity on T2-weighted images. Variable signal intensity may be due to necrosis, hematoma, or fat. After administration of contrast, the inhomogeneity of the tumor increases. A sharp demarcation of tumor tissue and renal parenchyma can be seen as a pseudocapsule which appears hypointense on T2-weighted sequences. As with CT, local tumor staging has proved difficult with MRI. Lymph node metastases can be difficult to differentiate from reactive changes. Tumor invasion into the renal vein or IVC can be seen on T2 imaging as a high signal surrounded as a filling defect on post-contrast T1 imaging [9].
Nephroblastomatosis
Etiology and Presentation
Nephrogenic rests refer to the persistence of fetal renal embryonic tissue beyond 36 weeks of gestation. It is present in approximately 1 % of infant kidneys at autopsy [10]. The presence of diffuse or multiple nephrogenic rests is termed nephroblastomatosis [10]. Most nephrogenic rests undergo spontaneous involution; however, malignant transformation to Wilms’ tumor represents approximately 30–40 % of Wilms’ tumor precursor lesions [10].
Nephrogenic rests may occur in different areas of the kidney, intralobar or perilobar. Intralobar nephrogenic rests occur less commonly than the perilobar type but are more often associated with development of Wilms’ tumor [10]. Intralobar nephrogenic rests present with neoplastic changes at a younger age compared to the perilobar type (median 16 vs. 36 months) [10]. Intralobar nephrogenic rests are typically found in conjunction with Wilms’ tumor on imaging, while perilobar nephrogenic rests are more often diffuse presenting as unilateral or bilateral flank masses [10].
Perilobar nephrogenic rests are linked to abnormalities in WT2 and may be associated with Beckwith-Wiedemann syndrome and hemihypertrophy. Intralobar nephrogenic rests are linked to abnormalities in WT1 and may be associated with WAGR, Denys-Drash syndrome, and sporadic aniridia. Almost 100 % of patients with sporadic aniridia have intralobar nephrogenic rests and 20 % have perilobar rests [10].
Histopathology
Nephrogenic rests are less than 3 cm in diameter and appear as tan nodules within the normal renal parenchyma. In the diffuse form, nephroblastomatosis appears as white plaques replacing much of the renal parenchyma or may form a discrete rind at the kidney periphery. Perilobar nephrogenic rests are found only in the cortex or at the corticomedullary junction. The margins are irregular and indistinct and may interdigitate with normal adjacent renal interstitium. Perilobar rests are composed predominantly of blast cells. Intralobar rests, in contrast, can be located anywhere within the renal parenchyma, and the predominant cell type is either stromal or epithelial [10].
Imaging
Nephroblastomatosis appears on imaging as discrete foci or as diffuse areas of the kidney [11]. On ultrasound, foci appear as homogeneously isoechoic or slightly hypoechoic areas compared to the normal renal cortex. In the diffuse form, nephroblastomatosis appears as a peripheral rim that is homogenous and hypoechogenic compared to the normal renal parenchyma. Because of the similar echogenicities of nephrogenic rests and renal cortex, it may be difficult to demonstrate nephrogenic rests smaller than 1–2 cm in diameter on ultrasound [11].
On unenhanced CT, nephrogenic foci appear as isodense or slightly hyperdense areas compared to renal cortex. Small lesions are nearly impossible to identify. After contrast administration, foci appear as homogenous, hypodense lesions because of poor contrast uptake [11]. Isolated nephrogenic rests have a nodular appearance and enhance less than the adjacent renal parenchyma. If numerous, they may cause the surface of the kidney to appear lobulated [10]. In the diffuse form, the surrounding rim of nephroblastomatosis appears as homogenously hypodense areas compared to the renal cortex (Figs. 11.3 and 11.4) [11].
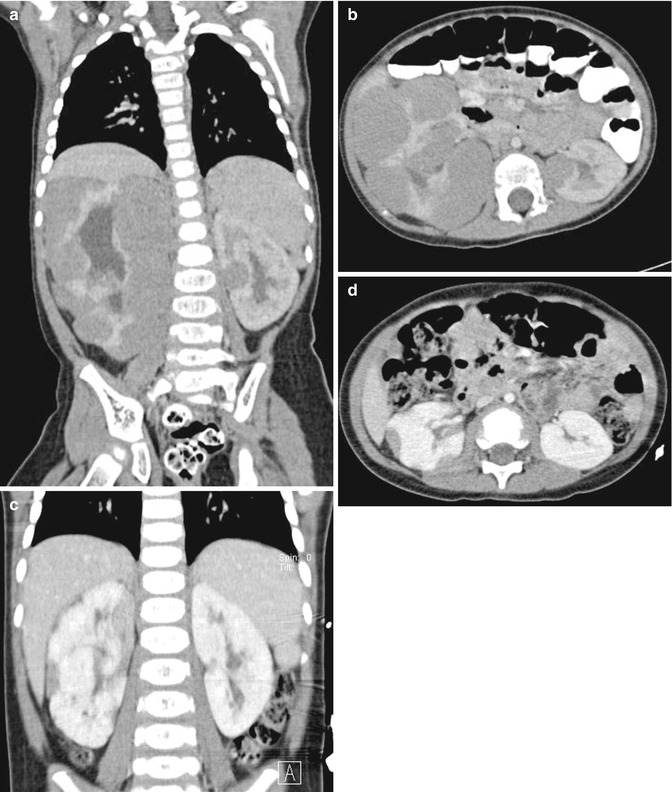
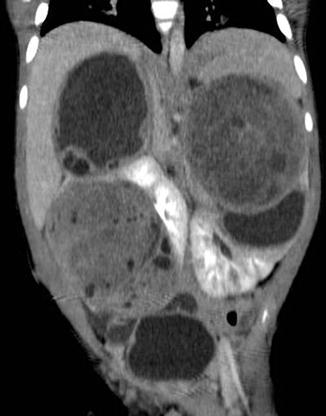
Fig. 11.3
Nephroblastomatosis. CT abdomen, axial and coronal, before (a and b) and after (c and d) chemotherapy. Peripheral lobular homogeneous hypodense rim of soft tissue masses in the right kidney, with enlargement of the right kidney and pelvicaliectasis. Solitaire small round soft tissue mass mid left renal pole. Successful response to chemotherapy with interval reduction of tumor bulk in bilateral kidneys
Fig. 11.4
Nephroblastomatosis with bilateral Wilms’ tumor. CT abdomen, coronal excretory phase: Several large masses arising off both kidneys, some with increased heterogeneous soft tissue components compatible with Wilms’ tumors
On MRI, nephrogenic foci appear as isointense or slightly hypointense to the renal cortex on T1- and T2-weighted images. After gadolinium enhancement, the lesions remain homogeneously hypointense compared to the bright enhancing renal cortex [11].
In differentiating Wilms’ tumor from nephroblastomatosis, the latter demonstrates near uniform homogeneity, while the former appears heterogenous on CT and MRI imaging especially after contrast or gadolinium administration (Fig. 11.5) [10–12].
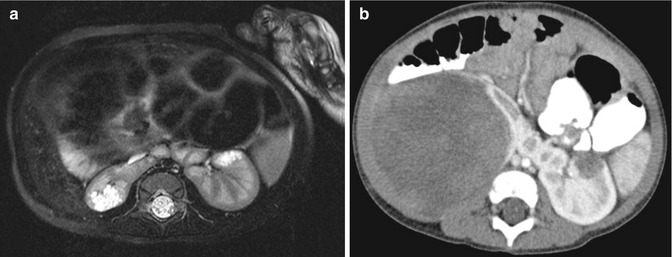
Fig. 11.5
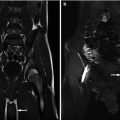
Nephroblastomatosis. MRI abdomen, axial T2 fat saturation with high resolution (a) and CT abdomen, corticomedullary phase 7 months later (b). Focal perilobar multilocular T2 hyperintense cystic mass in the right moiety of a horseshoe kidney, with interval replacement and development of large soft tissue tumor arising from the same location compatible with malignant transformation of a nephroblastic rest into a Wilms’ tumor
Renal Medullary Carcinoma
Etiology and Presentation
Renal medullary carcinoma (RMC) is a rare and fatal pediatric renal tumor that typically affects children of African descent with the sickle cell trait or SC disease. Patients present at 11–39 years with a 3:1 male predilection between 11 and 24 years and then without gender predilection subsequently. RMC most commonly presents as hematuria and less commonly as flank or abdominal pain or a palpable abdominal mass. Approximately 70 % of lesions occur on the right side [13]. The lesion is thought to arise from the collecting system and it grows to invade the surrounding vascular and lymphatic structures [14]. Metastatic disease to the liver, lung, and omentum is often present at diagnosis. Mean time to survival after diagnosis is approximately 15 weeks [15].
Histopathology
Grossly, RMC is a lobulated, tan, firm, or rubbery mass with variable degrees of hemorrhage and necrosis. The lesion has a dominant medullary location and extends into a large portion of the renal parenchyma. Invasion into the surrounding calyces or pelvis, vascular, lymphatic, and perinephric tissue is often present. Microscopically, RMC demonstrates stromal desmoplasia and inflammation. Satellite lesions are present throughout the renal cortex and pelvic soft tissue. Cells have a dark cytoplasm, clear nuclei, and prominent nucleoli. Hemorrhage and necrosis are often present.
Imaging
There are only a few reported cases of ultrasound findings for RMC given its rarity [13, 16, 17]. Sonogram may detect a heterogenous, hyperechoic renal mass [17] occupying the renal pelvis and most of the renal cortex [16]; however, others have found ultrasound to be unreliable [13].
CT imaging demonstrates an infiltrative, centrally located tumor with caliectasis. Low attenuation areas correspond to areas of necrosis. Hemorrhage, retroperitoneal adenopathy, and metastases are often present. After administration of contrast, renal medullar carcinoma demonstrates heterogeneous contrast enhancement with preservation of the surrounding renal contour.
Clear Cell Sarcoma of the Kidney
Etiology and Presentation
Clear cell sarcoma of the kidney is classified as an “unfavorable histology” tumor in the National Wilms’ Tumor Study Group (NWTSG) [18]. Kidd recognized it as a distinct entity and also noted its proclivity to metastasize to bone [18]. Clear cell sarcoma of the kidney may present as a palpable abdominal mass, as hematuria, or as increasing abdominal girth [19]. Approximately 50 % of cases are diagnosed between years 2 and 3 of life. Mean age at diagnosis is 36 months (range 2 months–14 years) [4]. It shows a 2:1 male to female predominance [4].
Approximately 25 % of patients present with stage I disease, 37 % present as stage II disease, 34 % present as stage III disease, and 4 % present with distant metastases [4]. There are no reported incidences of bilateral disease. Ipsilateral renal hilar lymph node metastases are the most common site of advanced disease at presentation occurring in up to 29 % [4]. Bone metastases are the most common site of disease relapse, but other sites include the lung, abdomen, retroperitoneum, brain, and liver [4]. Long-term follow-up is warranted as approximately 20 % of patients will present with metastases 3 years or more after time of diagnosis. Prognostically, four independent variables affect overall survival: stage at presentation (stage 1 >98 %), treatment with doxorubicin, patient age, and the absence of necrosis [4].
Histopathology
Grossly, clear cell sarcomas are large, tan-gray, soft, mucoid unicentric tumors that distort or nearly completely replace the kidney [4]. They have a well-circumscribed appearance with sharp kidney-tumor borders. Necrosis and hemorrhage are commonly present [4]. Cystic foci are present in nearly all cases. Mean size of tumor is 11.3 cm (range 2.3–24 cm) and mostly the common site of origin is the renal medulla. Microscopically, the classic pattern can be described as cords of plump cells separated by fibrovascular septa, with indistinct cell borders and open chromatin. Cord cells are loosely spaced and separated by extracellular mucopolysaccharide matrix. In addition to the classic appearance, many tumors demonstrate a second morphology that may seamlessly blend with the classic pattern [4].
Imaging
Radiologically, clear cell sarcoma of the kidney appears as a focal mass with a dominant soft tissue component with areas of necrosis [19]. Ultrasonography demonstrates an inhomogeneous pattern of soft tissue echoes and echo-poor areas that correspond to necrosis. Multiple anechoic areas correspond to septated cystic spaces. This may account for the bulk of the tumor and may make differentiation from multilocular cystic nephroma or cystic Wilms’ variant difficult [19]. Specular echoes corresponding to calcifications may also be seen.
After contrast administration, CT imaging demonstrates a heterogeneous tumor with attenuation less than that of normal renal parenchyma. Necrotic areas are clearly evident and calcifications or hemorrhage may also be demonstrated [19].
Rhabdoid Tumor of the Kidney
Etiology and Presentation
Rhabdoid tumor of the kidney was originally categorized as an unfavorable histological subtype of Wilms’ tumor [6] but was separated out as a distinct entity in 1998 [20]. Malignant rhabdoid tumor of the kidney is one of the most aggressive pediatric renal tumors and accounts for approximately 2 % of all pediatric renal tumors with 80 % occurring under the age of 2 years [21].
These tumors present as a palpable abdominal mass, hematuria, or fever [22] and sometimes with hypercalcemia [20]. Median age at diagnosis is 11 months and occurs more frequently in males (3:2). Extrarenal extension is commonly seen [23] and those presenting with metastases (lung and liver) die of disease progression [4]. Rhabdoid tumors are associated (up to 10 %) with the development of synchronous or metachronous primary tumors of the central nervous system [23]. Most brain lesions are reported as medulloblastoma, but other diagnoses include primitive neuroectodermal tumors, ependymoma, or gliomas [20].
Histopathology
Grossly, rhabdoid tumors resemble a bulky mass, largely replacing the kidney with preservation of a thin rim of peripheral renal tissue. Well-circumscribed boundaries [23] are not commonly seen and they frequently invade [23]. Mean tumor size at time of diagnosis is approximately 9.6 cm (range 3–17 cm). The tumor is soft, friable, gray to pink tan in color with focal areas of necrosis and hemorrhage. The tumor tends to occupy the central portion of the kidney with involvement of the renal hilar structures. The renal vein is often filled with tumor, and involvement of the renal pelvis occurs in some. Most specimens show capsular invasion, intrarenal and intrapelvic involvement, and infiltration of the renal sinus.
Microscopically, rhabdoid tumor of the kidney shows a wide range of morphological diversity. Rhabdoid cells are large with centrally placed nucleoli and abundant cytoplasm with prominent cytoplasmic inclusions [20]. The classical variant shows sheets of solid large, ovoid to polygonal cells, with abundant eosinophilic cytoplasm, intracytoplasmic inclusions, and conspicuous nucleoli [20].
Imaging
CT imaging demonstrates a heterogeneous, lobular, hilar tumor with central hypodense areas corresponding to necrosis and hemorrhage (Fig. 11.6) [22]. Calcification appears linearly and tends to outline the tumor lobule. Crescent-shaped hypodense areas in the periphery of the kidney outline tumor nodules and correspond pathologically to subcapsular hematoma. CT often demonstrates extension into adjacent structures such as the renal vein and inferior vena cava. Distant metastases to the lung may also be present on preoperative imaging. Imaging of the head sometimes demonstrates a synchronous CNS lesion which is not commonly seen in other pediatric renal tumors.
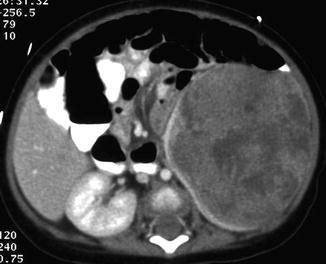
Fig. 11.6
Rhabdoid tumor. CT abdomen, axial late nephrographic phase: large heterogeneously enhancing mass in the left kidney (claw sign) with central necrosis and possible hemorrhage (Courtesy of Dr. Richard D. Bellah, MD at Children’s Hospital of Philadelphia)
Renal Cell Carcinoma
Etiology and Presentation
Renal cell carcinoma (RCC) accounts for less than 4 % of pediatric renal tumors [24]. The median age at diagnosis is 9–12 years [25] without gender predilection [26]. Most patients present asymptomatically [25, 26]. Symptomatic patients may have hematuria, flank pain, or a palpable abdominal mass. The overall survival rate for children nears 50–60 % but approaches 90 % for those with lower tumor stage. Metastases (lungs, bones, liver, or brain) occur in approximately 20 % [14] and demonstrate survival of approximately 10–15 % at 5 years [25].
Whereas the von Hippel-Lindau (VHL) gene is implicated both in familial and sporadic cases of clear cell RCC in adults, recent reports suggest that this mutation is largely absent in children [25]. Translocation carcinomas have recently emerged as the predominant subtype of renal cell carcinoma in children. One such translocation t(X;17)(p11.2;q25) leads to a fusion of the transcription factor gene TFE3 with the ASPL gene on 17q25. Another translocation t(X;1)(p11.2;q21) leads to a fusion TFE3 transcription factor gene on Xp11.2 with the PRCC gene at 1q21.2.
Histopathology
Histologically, RCC are characterized into clear cell, papillary, and chromophobe carcinoma, as in adults. Most tumors are surrounded by a pseudocapsule and angioinvasion is common. Microscopically, translocation carcinomas contain tumor cells resembling conventional clear cell carcinoma but may have areas of papillary features, granular eosinophilic cytoplasm, and calcifications [26]. The architecture may vary but is predominantly solid, tubular, acinar, or alveolar with a hyalinized stroma demonstrating focal inflammatory infiltrates. Cells have voluminous clear cytoplasms with bulging cell borders [24]. Nuclei are vesicular, moderately pleomorphic, and wrinkled [24].
Imaging
RCC commonly appears as large, heterogenous enhancing masses on CT imaging. Many tumors show internal or adjacent hemorrhage, internal calcifications, and neovascularity. On MRI, tumors commonly demonstrate heterogeneous enhancement and intratumoral hemorrhage.
Congenital Mesoblastic Nephroma
Etiology and Presentation
Congenital mesoblastic nephroma (CMN) is the most common pediatric renal tumor diagnosed within the first 3 months of age [26], and roughly 90 % are diagnosed within the first year of life. The tumor shows a male to female predominance of 2:1 [26]. CMN most commonly presents as a palpable abdominal mass, gross hematuria, or as an incidental finding on prenatal sonography. Hypertension and hypercalcemia may occur.
Two morphologic variants exist: classic and cellular. The classical type carries a better prognosis, while the cellular variant has been shown to invade perirenal fat, adjacent organs, and the renal vein and has also been shown to recur locally [27]. Mixed variants, composed of elements of classic and cellular, have also been described. The classic variant is usually diagnosed in the neonatal period, while the cellular variant presents at a later date and is typically larger [28].
Histopathology
Grossly, the classic variant appears as a solid, firm, light yellow mass with no capsule and poorly defined margins, resembling uterine leimyoma [14, 26]. The cellular variant tends to be soft and fleshy, with areas of hemorrhage or necrosis [28]. Other aggressive features of the cellular variant include invasion of surrounding structures such as the colon, small bowel, and pancreas, vascular invasion, tumor thrombus, and extension into the perihilar connective tissue [28].
The classic variant can be described as fibroblastic spindle cells arranged in bundles and fascicles that infiltrate the renal parenchyma [26]. There is low cellularity and atypia is absent [28]. The cellular variant is characterized by increased cellularity, vacuolated cytoplasm [28], plump cells with increased nuclear pleomorphism, a high nuclear/cytoplasmic ratio, and frequent mitotic figures [27]. The tumor cells are larger, fusiform to ovoid, and arranged in sheets with less well-defined bundles [28]. The tumor cells may be surrounded by a highly vascular hyalinizing stroma [28].
Imaging
On prenatal ultrasound, CMN can appear as an enlargement of the kidney [29] or as a large heterogeneous echogenic mass [30] that can sometimes be difficult to discern from the normal renal parenchyma [29] (Fig. 11.7). It is commonly associated with fetal hydrops and polyhydramnios [29]. After birth, CMN appears as a mass with a central poor echoic area surrounded by a thin echogenic rim (ring sign) [28]. This corresponds pathologically to dilated blood vessels at the periphery of the tumor [27] (Fig. 11.8a).
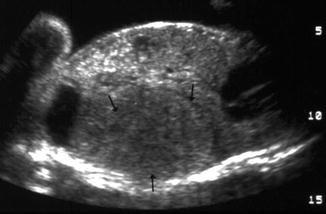
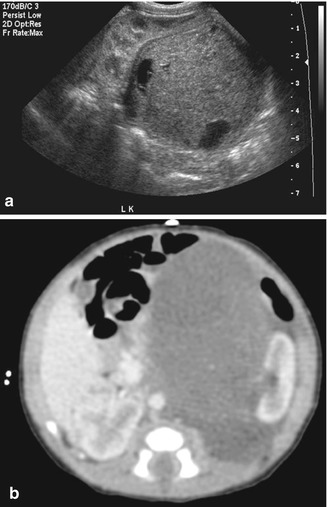
Fig. 11.7
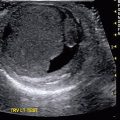
Mesoblastic nephroma. Fetal ultrasound. Large hypo- to isoechoic mass in the renal bed (Courtesy of Dr. Richard D. Bellah, MD at Children’s Hospital of Philadelphia)
Fig. 11.8
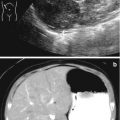
Mesoblastic nephroma. Grayscale ultrasound (a) large hypoechoic mass in the left renal bed, with peripheral cystic areas, possibly representing blood vessels and/or necrosis. CT abdomen (b), axial corticomedullary phase. CT demonstrates homogeneous enhancement of the left hilar mass distorting and lateral displacing the left kidney
On nonenhanced CT, CMN appears as a large heterogeneous mass with areas of high attenuation which correspond to areas of hemorrhage. After contrast administration, CT demonstrates a heterogeneous mass with enhancement of the solid component and areas of cystic and necrotic changes (Fig. 11.8b). Pathologically, the cellular variant is more likely to be identified if preoperative CT reveals cystic and hemorrhagic areas, while the classic variant is more likely to be a large, uniform, soft tissue mass [27].
MRI before gadolinium administration shows solid components with low signal intensity on T1 and high signal intensity on T2 [27]. High signal intensity on T1 corresponds to areas of hemorrhage. After administration of gadolinium, MRI demonstrates variable heterogeneous enhancement of the solid components.
Multilocular Cystic Renal Tumor
Etiology and Presentation
Multilocular cystic renal tumor refers to two entities that cannot be distinguished on the basis of gross pathology or radiologic findings. Multilocular cystic renal tumor is a benign lesion composed of multiple noncommunicating cysts separated by septa devoid of any blastemal or other embryonal elements. Partially differentiated cystic nephroblastoma refers to a predominant cystic lesion where the septa contain blastemal or other embryonal elements. Multilocular cystic partially differentiated nephroblastoma is typically a benign lesion; however, local recurrence after nephrectomy may occur. The etiology is unclear, but there are several hypotheses. If there are associated nodules of tumorous tissues, the multicystic lesion should be considered malignant. The prognosis for these malignancies is better than their corresponding non-cystic tumors (i.e., Wilms’ tumor and RCC).
Multilocular cystic renal tumor presents most commonly as a painless abdominal mass [31]. It shows a bimodal distribution (3 and 25 years) affecting males more commonly than females during childhood, with a ratio of 2:1 and 8:1 in adults.
Histopathology
Grossly, multilocular cystic renal tumor can be described as a solitary, well-circumscribed multiseptated renal lesion containing noncommunicating fluid-filled loculi surrounded by a thick capsule (rind-like) compressing a thin rim of renal parenchyma. Both the capsule and the septations may be thick. The actual cystic spaces may not be very large. Grossly, the lesions may range in size from several millimeters to several centimeters. Mean size is 9.7 cm. The malignant spectrum of this cystic mass tends to be much larger and may be as big as 20 cm. Calcifications may be seen within the septa or mass wall. Hemorrhage, necrosis, and calcification are uncommon.
Microscopically, in multilocular cystic nephroma, the cysts are lined by flattened, cuboidal, or hobnail epithelium. In the well-differentiated, benign subtype, the septa consist of fibrous tissue without blastemal elements. In contrast, the septa in cystic partially differentiated nephroblastoma contain blastemal cells.
Imaging
Multilocular cystic nephroma and multilocular partially differentiated cystic nephroblastoma cannot be distinguished radiographically. On ultrasound, the kidney appears as a multiloculated mass with thin septa. The renal parenchyma can be compressed by the renal mass forming a thin rim of parenchyma (beak sign).
On nonenhanced CT, multilocular cystic renal tumors appear as sharply circumscribed, multiseptated renal mass. Occasionally, the mass may appear solid when the cysts come in close approximation. The interior of the cysts show attenuation that is consistent with water. With contrast, the septations may enhance; however, the contrast does not accumulate within the individual cysts (Fig. 11.9).
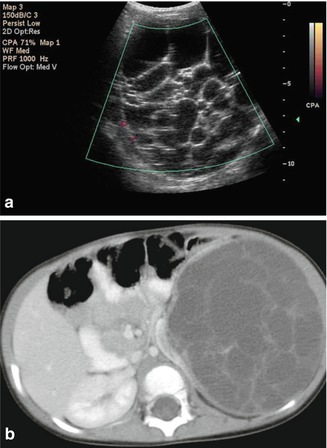
Fig. 11.9
Multilocular cystic renal tumor (1.5-year-old male). Color Doppler ultrasound (a) and axial CT (b) late nephrographic phase. Large multilocular cystic lesion, without internal vascularity. Lesion resulting in distortion and splaying of the left kidney. No accumulation of contrast on delayed imaging (not shown)
On MRI, the septa and renal capsule appear as low signal intensity on T1-weighted images. The septa enhance after gadolinium administration. On T2-weighted images, the fluid contents of the cyst display high signal intensity (Fig. 11.10).
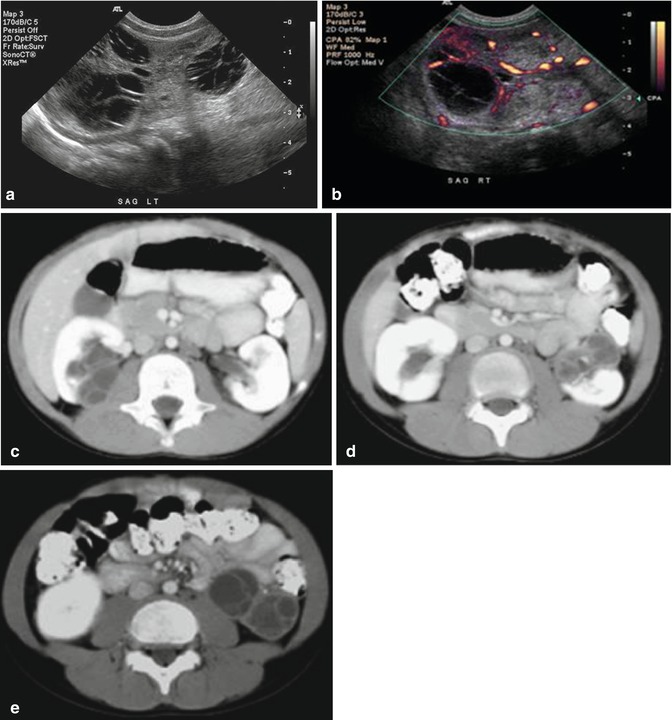
Fig. 11.10
Multilocular cystic renal tumor. Grayscale (a) and power Doppler (b) ultrasound kidneys. Several multilocular cystic lesions in both kidneys, at least three in the left, with thin avascular septations. CT abdomen, sequential axial images in nephrographic phase. (c, d, and e) Bilateral multilocular cystic masses, with herniation of the right lesion into the renal pelvis
Metanephric Adenoma
Etiology and Presentation
Metanephric adenoma is typically a benign renal lesion presenting as an incidental, usually unilateral finding on imaging [32, 33]. If symptomatic, metanephric adenoma may present as a palpable mass, abdominal or flank pain, or hematuria [32]. An association with polycythemia is reported in approximately 10 % of cases [34]. Metanephric adenoma most commonly occurs in females with a mean age of 41 (range 5–83 years). It demonstrates a female to male ratio of approximately 2:1 [32].
Histopathology
Grossly, metanephric adenoma appears as a well-circumscribed, solid, gray to tan to yellow tumor that is soft to firm [32–34]. Tumors may contain areas of hemorrhage, necrosis, or cystic degeneration [34]. Calcification may occur scattered throughout the tumors, in the cyst walls, or in the center of the tumor [32]. Most tumors are surrounded by a distinct capsule; however, some demonstrate a discontinuous or absent capsule [32].
Microscopically, these tumors are composed predominantly of small uniform epithelial cells separated by an acellular stroma [32, 34]. In about 50 % of cases, papillary structures are observed which consist of polypoid fronds or papillary infoldings forming a glomeruloid appearance [34] and are frequently calcified [32]. Cells demonstrate little cytoplasm with irregular rounded or ovoid nuclei with delicate chromatin [32, 34]. Nucleoli are not prominent and pleomorphism and mitotic figures are typically absent [32, 34].
Imaging
On ultrasound, metanephric adenomas appear as hypo-, iso-, or hyperechoic tumors compared to adjacent renal cortex. Color Doppler reveals no vascular flow in the tumor [33].
On unenhanced CT, tumors demonstrate higher attenuation than the adjacent renal parenchyma. With the addition of contrast enhancement, tumors typically demonstrate a lower attenuation than the renal parenchyma reflecting the hypovascular nature of these tumors. Areas of calcifications can frequently be seen.
On MRI, both T1- and T2-weighted images demonstrate lower signal intensity than the renal parenchyma even with the addition of gadolinium (Fig. 11.11).
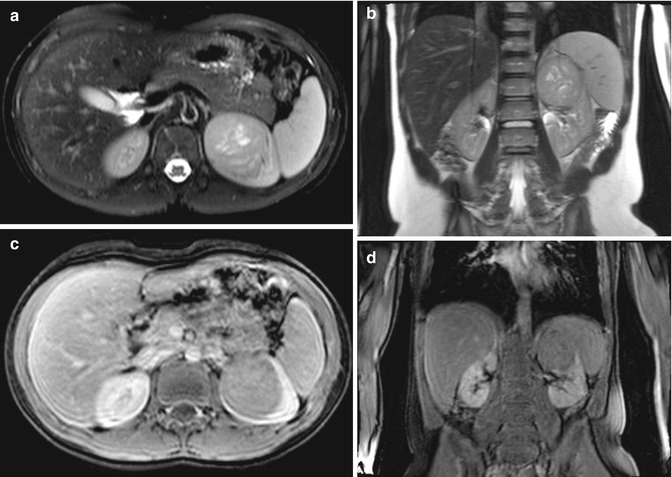
Fig. 11.11
Metanephric adenoma. MRI abdomen, axial T2 fat saturated (a), coronal T2 (b), axial (c), and coronal (d) T1 GRE fat saturated post contrast. Large heterogeneous T2 isointense masses in renal cortex arising from the upper pole of the left kidney, with central cystic components. Decreased enhancement relative to remaining kidney post contrast
Angiomyolipoma
Etiology and Presentation
Renal angiomyolipomas (AML) are benign neoplasms composed of adipose tissue, thick-walled blood vessels, and smooth muscle in varying amounts. The arteries in AMLs are deficient in the internal elastic membrane and are consequently torturous and prone to aneurysmal formation and rupture [35]. Approximately 10 % of patients with AML are associated with tuberous sclerosis complex (TS), and up to 80 % of patients with TS develop AML. Deletions of tumor suppressor genes located on chromosomes 9 (TSC1) and 16 (TSC2) are believed to cause AML growth.
Patients with TS are more likely to have numerous bilateral tumors that present at an earlier age and which exhibit increasing size with age [36, 37]. Patients with TS demonstrate three renal phenotypes: AML, renal cysts, and renal carcinoma with AML being the most common phenotype [38]. Many patients with AMLs less than 4 cm in size present incidentally on imaging [35, 39, 40]. Lesions larger than 4 cm commonly present with acute flank or abdominal pain, palpable mass, or hematuria [39]. In some cases, patients may present with Wunderlich syndrome, shock secondary to retroperitoneal bleeding [38]. AMLs are believed to be prone to rupture secondary to the dysplastic elastic tissue leading to aneurysmal formation [14].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


