CHAPTER 33 Sellar and Juxtasellar Tumors
The sellar region is defined by the sella turcica and its contents. The parasellar region has no precise boundaries but is considered to be the area surrounding the sella turcica, including the cavernous sinus. Many diverse lesions involve the sellar and juxtasellar regions, including primary benign and malignant neoplasms, metastases, perineurial spread of tumor, and direct invasion from adjacent tumors. The most common lesions are pituitary macroadenomas, craniopharyngiomas, meningiomas, metastasis, and hypothalamic-optic gliomas, which together account for about 75% of lesions in this area.1 Aneurysms clearly must be considered in the differential diagnosis of this region but are discussed elsewhere.
PITUITARY ADENOMA
Epidemiology
The incidence of pituitary adenomas ranges from 2% to 27% in the general population. They occur with greater frequency in females than males and most commonly arise in patients between 20 and 50 years of age.1 Adenomas are the most common of the sellar lesions, accounting for up to 90% of neoplasms that involve this region.2
Clinical Presentation
Microadenomas frequently come to clinical attention because of hormonal secretion. Prolactin is the hormone most frequently elevated (25%) and can result in infertility, amenorrhea, and galactorrhea in women and decreased libido and impotence in men. Growth hormone–secreting adenomas result in acromegaly in adults and gigantism in children, whereas adrenocorticotropic hormone (ACTH)–secreting adenomas arising from the posterior pituitary or the neurohypophysial region cause Cushing’s syndrome. Thyroid-stimulating hormone (TSH) and folliclestimulating hormone/luteinizing hormone (FSH/LH)–secreting adenomas are rare. Twenty-five to 30 percent of pituitary microadenomas are nonfunctional.1
Macroadenomas frequently do not elaborate hormones. Instead, they present at larger size with mass effect on the chiasm causing visual impairment, compression of the pituitary gland and/or stalk causing pituitary insufficiency, compression of the third ventricle and foramina of Monro causing hydrocephalus, or invasion of the cavernous sinus causing cranial nerve palsies. In males, prolactinomas may reach large size, because the symptoms are less readily identifiable than in females. Occasionally, mass effect on the infundibulum causes elevated prolactin levels, but in these cases the prolactin levels typically do not exceed 150 ng/mL. Overall, about 40% of macroadenomas invade the cavernous sinus to some extent but rarely cause cranial nerve palsy.2
Intratumoral hemorrhage can occur in up to 15% of adenomas (Fig. 33-1). Both microadenomas and macroadenomas can undergo a rapid increase in size in the event of pituitary apoplexy, which is an acute hemorrhage or infarction of a pituitary adenoma. Apoplexy results in acute symptoms, such as headache, vomiting, ocular motility disturbance, and possibly a sudden decrease in vision.3 Seizures or decreased consciousness may also occur.
Pathophysiology
Pituitary adenomas arise from the adenohypophysis, or anterior pituitary gland. Traditional embryology describes the anterior pituitary as arising by invagination of the rostral stomadeum.4 The pars distalis makes up the majority of the anterior lobe. A portion of the pars distalis also extends superiorly and contributes to the anterior aspect of the infundibulum. Therefore, the infundibulum is composed of components from both the anterior and posterior pituitary lobes, explaining the occurrence of occasional adenomas along the stalk.4
Pathology
Microscopically, adenomas are composed of sheets of monomorphic cells. Prolactinomas may show psammomatous calcifications and amyloid deposits. Patients with pituitary apoplexy have hemorrhage and necrosis within the gland.
Imaging
CT
The appearance of macroadenomas varies with the size of the lesion. Nearly all (94%-100%) macroadenomas enlarge the sella turcica, but sellar enlargement is not specific for adenomas. Sellar enlargement has also been reported in more than 50% of nonadenomatous lesions involving the sella turcica, including meningiomas, craniopharyngiomas, and Rathke’s cleft cysts.5 On contrast-enhanced images, macroadenomas may appear isodense or hypodense as compared with the cavernous sinus. The imaging characteristics will also depend on the degree of necrosis and hemorrhage.
MRI
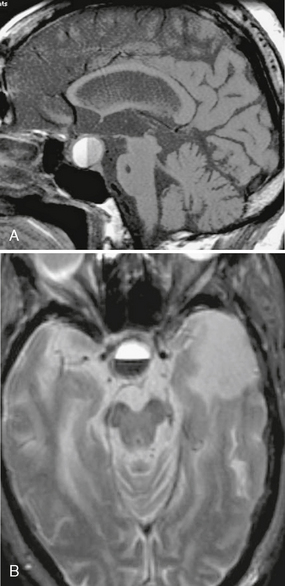
MRI can accurately demonstrate microadenomas as small as 3 mm and frequently succeeds in delineating even smaller lesions. Coronal and sagittal imaging are the most valuable for assessing microadenomas and macroadenomas. They are especially helpful for assessing cavernous sinus invasion and involvement of the optic nerves by macroadenomas. On T1-weighted (T1W) imaging, microadenomas are hypointense to isointense in comparison with the normal gland. They may deviate the infundibulum and bow the superior aspect of the gland superiorly. However, caution should be used when evaluating the tilt of the infundibulum, because some normal patients have infundibular deviation due to ectopic insertion of the infundibulum or eccentric gland position. Hemorrhage into the adenoma may cause T1 shortening. Suspected microadenomas may be evaluated by dynamic imaging, because they will enhance more slowly than the surrounding gland and therefore can be differentiated from the gland more easily (Fig. 33-2). On delayed contrast-enhanced MRI, the microadenomas will enhance like the gland itself and become difficult to distinguish. Still later, continued enhancement of the adenomas and washout of contrast from the gland may reverse the expected appearance, so the normal (now nonenhancing) gland is mistaken for the adenoma.
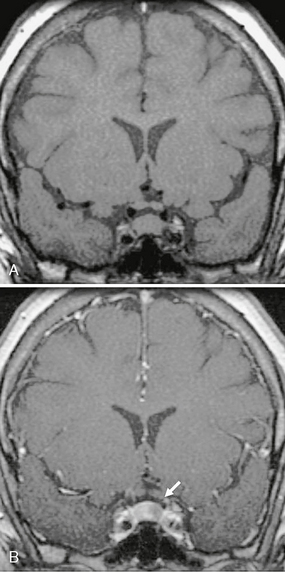
FIGURE 33-2 Pituitary microadenoma. Coronal T1W MR image without contrast (A) and early postcontrast T1W MR image (B) demonstrate a 7-mm focus of nonenhancement (arrow) consistent with a pituitary microadenoma.
Macroadenomas are variable in their MRI appearance depending on the degree of necrosis and hemorrhage. They usually appear isointense to gray matter on T1W and T2-weighted (T2W) imaging, are hyperintense on fluid-attenuated inversion recovery (FLAIR) images, and show nearly constant heterogeneous contrast enhancement. They often show areas of cyst and hemorrhage, especially in the larger tumors. Coronal T2W images typically display the optic chiasm most clearly as a crescent of low signal crossing above the sella. The paired A1 segments of the anterior cerebral arteries pass immediately above the prechiasmal optic nerves or the chiasm itself. Compression of the chiasm by macroadenoma typically appears as elevation and thinning of the low signal “chiasmal crescent” with elevation of the flow voids of the A1 segments above it. Accurate detection of extension to the cavernous sinus is difficult. MRI is only 55% sensitive for invasion of the cavernous sinus. Invasion can be suspected when at least 50% of the cavernous segment of the internal carotid artery is surrounded by the macroadenoma. Unilateral carotid artery encasement is the most specific imaging sign of cavernous sinus involvement (Fig. 33-3A).6 Characteristically, pituitary macroadenomas do not narrow the carotid artery, even when they encase it, whereas meningiomas of the cavernous sinus do narrow the carotid artery when they surround it. Therefore, preservation of normal caliber of an encased internal carotid artery suggests that the cavernous sinus has been invaded by a macroadenoma, rather than meningioma. This sign, however, is imperfect. Macroadenomas can also erode through the floor of the sella turcica. Large lesions may extend into the sphenoidal sinus and then through the sphenoidal sinus into the nasopharynx, mimicking carcinoma of the sphenoidal sinus or nasopharynx (see Fig. 33-3B). Because nasopharyngeal carcinomas also tend to narrow the internal carotid arteries, preservation of the caliber of these vessels again suggests pituitary macroadenoma. MR spectroscopy may show a choline peak or no metabolites at all.7
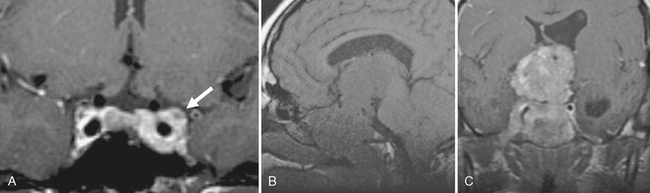
FIGURE 33-3 Invasive pituitary macroadenoma. Coronal T1W MR image (A) after gadolinium administration demonstrates extension of enhancing lesion around the left cavernous internal carotid artery (arrow). Sagittal T1W precontrast (B) and coronal T1W postcontrast (C) MR images of another patient demonstrate a large heterogeneously enhancing mass that expands the sella and extends inferiorly to involve the sphenoidal and ethmoidal air cells. No arterial narrowing is noted despite the large size of the lesion. However, the lesion results in hydrocephalus with enlargement of the left lateral ventricle.
PITUITARY CARCINOMA
Epidemiology
Pituitary carcinomas comprise 0.2% of adenohypophysial neoplasms. Approximately 140 cases have been reported thus far. Pituitary carcinomas are equally frequent in males and females. The mean age at presentation is 44 years.8 The diagnosis of pituitary carcinoma requires documentation of metastases. Patients typically survive about 4 years after lesion discovery.
Clinical Presentation
Pituitary carcinoma may first present as an invasive pituitary macroadenoma, only to demonstrate craniospinal or systemic metastases after a latent period of 5 to 10 years. The most common sites of hematogenous spread are liver and bone. Patients with craniospinal metastases tend to have a better prognosis than those with systemic spread. Pituitary carcinomas may secrete hormones, leading to endocrine imbalance. Their prognosis is then related to the endocrinologic subtype, with a worse prognosis for those secreting ACTH.9
Pathophysiology
Pituitary carcinomas may result from malignant transformation of benign adenomas. Chromosomal gains, especially additions to 14q, are likely involved with the malignant degeneration. In addition, pituitary carcinomas have reduced expression of the protein nm23, which normally prevents progression of the cell cycle and reduces the potential for metastasis.8
Like pituitary adenomas, pituitary carcinomas arise from adenohypophysial cells, so they can be difficult to differentiate from adenoma by histology. Features typical for malignancy, including nuclear pleomorphism, invasion, necrosis, and increased numbers of mitotic figures, are not reliable for distinguishing between the two.8 This diagnosis cannot be made unless there is evidence of distant metastases.
Imaging
CT and MRI
Both CT and MRI reveal a lesion that mimics invasive macroadenoma. The only distinction on imaging is the presence of metastasis (Fig. 33-4).
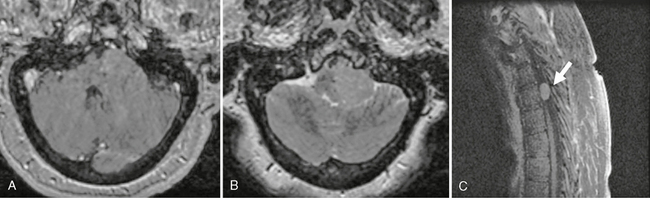
FIGURE 33-4 A 63-year-old woman presented with a history of pituitary adenoma resection 10 years previously. Follow-up imaging initially was negative, but on recent follow-up a left cerebellopontine angle mass was noted (A and B). Biopsy was consistent with pituitary carcinoma. Imaging of the spine revealed multiple enhancing lesions, the largest of which was in the thoracic region (C, arrow).
GRANULAR CELL TUMORS
Epidemiology
Granular cell tumors are the most common primary tumor of the posterior pituitary gland. They most frequently present in the fifth decade and are twice as common in women as men.10
Clinical Presentation
Granular cell tumors are hormonally silent, so they usually present as masses. Most patients have visual complaints from compression of the optic chiasm.10 Approximately 50% of patients show secondary signs of pituitary dysfunction, including hyperprolactinemia, which is presumed to result from compression of the pituitary stalk by the primary mass. Compression of the stalk impedes transport of pituitary-releasing hormones from the hypothalamus to the pituitary gland, leading to endocrine dysfunction (so-called stalk effect).
Pathophysiology
The cell of origin of granular cell tumors has not been identified definitively. Some evidence suggests that granular cell tumors originate from pituicytes. This is supported by the presence of glial fibrillary acidic protein (GFAP) within the tumor cells on electron microscopy. However, actual immunohistochemical staining for glial fibrillary acidic protein has been negative. There has been speculation that this may be explained by crowding of the cell cytoplasm by abundant lysosomes.10
Imaging
CT
On CT, granular cell tumors are supra- and intrasellar masses, which are iso- to hyperdense to gray matter and show contrast enhancement. These imaging findings are nonspecific and can resemble pituitary macroadenomas, meningiomas, craniopharyngiomas, and hypothalamic-chiasmatic gliomas. Calcification within the lesion has been reported but is not a typical imaging feature.10
MRI
On MRI, granular cell tumors have a nonspecific appearance. They are hypointense to isointense to gray matter on both T1W and T2W imaging.9 Granular cell tumors are vascular lesions, so MRI typically shows an enhancement within the sellar/suprasellar mass. Granular cell lesions do not cause significant edema in the surrounding brain. Their appearance is nonspecific, so imaging does not differentiate these tumors from other sellar/suprasellar lesions, including craniopharyngioma, pituitary adenoma, and meningioma. Absence of the posterior pituitary bright spot may provide a clue that the lesion originates from the posterior pituitary gland, but absence of the posterior pituitary bright spot on incidental MRI is another nonspecific sign present in up to 20% of normal subjects.9
PITUICYTOMA
Epidemiology
Pituicytomas are exceedingly rare. They occur from the third to the ninth decade and are more frequent in males.11
Pathology
Pituicytomas are highly vascular tumors. Complete surgical resection is difficult, so local recurrence is common after resection. These tumors are composed of spindle-shaped cells with round-to-oval nuclei and fibrillary cytoplasm. The cells align around the vasculature in a pattern similar to the normal neurohypophysial architecture. Pituicytomas do not demonstrate immunoreactivity for neuroendocrine markers or pituitary hormones.12 They do not exhibit Rosenthal fibers, microcysts, or granular bodies, so they are distinct from pilocytic astrocytomas.
Imaging
Special Procedures
Because of the highly vascular nature of pituicytomas, angiography is beneficial for surgical planning. Prominent arterial feeders can be seen to arise from the hypophysial arteries. There is no supply from the external carotid artery, which is helpful in distinguishing these lesions from meningiomas.12
CRANIOPHARYNGIOMA
Epidemiology
Craniopharyngiomas represent 1% to 3% of intracranial tumors. They are equally frequent in males and females. Craniopharyngiomas may present at any age but demonstrate clear bimodal age peaks, with special frequency first in childhood and then in the fifth decade. More than half of craniopharyngiomas present in children and young adults. Craniopharyngiomas are the most common intracranial neoplasm of nonglial origin in the pediatric population.13
Clinical Presentation
Craniopharyngiomas usually present as headache, nausea, and visual symptoms due to compression of the optic pathway. Hypothalamic dysfunction and pituitary dysfunction are common. Up to 80% of children show endocrine dysfunction at the time of presentation, most frequently growth disturbance.9 In adults, the most common presenting symptom is chronic headache.
Pathophysiology
The second theory is the metaplastic theory. The early depression that will lead to the mouth is called the stomodeum. The stomodeum contributes to the formation of the buccal mucosa. The metaplastic theory postulates that craniopharyngiomas arise from metaplasia of squamous epithelial cell rests, which are remnants of the area of the stomodeum that contributed to the buccal mucosa.14 The squamous papillary craniopharyngiomas, which are most common in adults, are thought to arise by this route.
Pathology
Pathologically, craniopharyngiomas are divided into adamantinomatous and squamous papillary variants. The adamantinomatous type resemble the enamel-forming neoplasms of the oropharynx. They tend to be more cystic, calcify more frequently, and are most common in children. The squamous papillary form is often more solid, less frequently calcified, and more frequent in adults. However, most tumors have mixed features. Attempts to correlate pathology with imaging and recurrence rate have not proved fruitful.15 Although craniopharyngiomas are histologically benign, slowly growing tumors, they are difficult to resect completely. They are often adherent to the pituitary stalk, the hypothalamus, the thalamoperforate vessels, and other small penetrating arteries and veins, so complete resection is difficult or impossible. For that reason they recur repeatedly, are locally aggressive, and may extend into the adjacent brain parenchyma. If the tumors are completely resected, recurrence is rare. With subtotal resection, however, only 47% of patients are disease free at 5 years.9 Craniopharyngiomas can recur locally, along the surgical tract, and even distant from the original site, suggesting cerebrospinal fluid (CSF) seeding.15
Imaging
CT
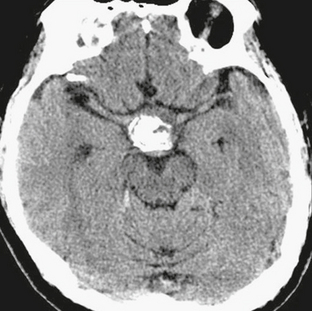
On CT, craniopharyngiomas are round lobulated masses centered within the suprasellar cistern and occasionally involving the sella. Cystic areas are frequent (up to 85% of craniopharyngiomas) and are variable in attenuation, depending on their cholesterol content. Calcification is more frequent in childhood craniopharyngiomas (up to 90%) than adult tumors (50%-70%) and tends to be more prominent and chunkier in children than adults.9 The calcification can be rim-like or conglomerate (Fig. 33-5). The solid portions of the tumor enhance markedly after contrast agent administration.
MRI
On MRI, the signal intensity of craniopharyngiomas is variable, depending on the cyst contents and degree of calcification. The cystic components may show T1 shortening. After contrast agent administration, craniopharyngiomas usually show enhancement in the solid portions and rim enhancement about the cysts (Fig. 33-6). MR spectroscopy has been reported to be useful in distinguishing craniopharyngiomas from hypothalamic astrocytomas and pituitary adenomas. Craniopharyngiomas demonstrate peaks from 1 to 2 ppm, which correspond to the region for lipid and lactate. Astrocytomas will demonstrate elevated N-acetyl-aspartate (NAA) to creatine ratios, and adenomas demonstrate no brain metabolites.
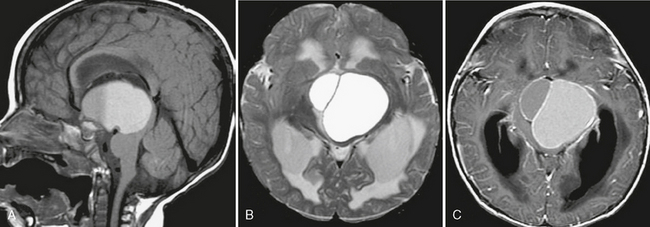
FIGURE 33-6 Craniopharyngioma. A, Sagittal T1W MR image demonstrates a large suprasellar mass with a fluid/fluid level. B, On the axial T2W MR image enlargement of the ventricles consistent with hydrocephalus as a result of the mass effect is better appreciated. T2 prolongation surrounding the ventricles is consistent with transependymal flow. C, On the postcontrast image, enhancement of the cyst walls is noted.
Craniopharyngiomas commonly cause T2 prolongation along the optic tracts. This sign is nonspecific and has been reported in other pituitary region tumors, including meningiomas and metastases.16 The high T2 signal may signify optic edema from compression by the tumor.
Special Procedures
Craniopharyngiomas are typically avascular on angiography and may be noted to encase or displace the vessels of the circle of Willis. Tumoral adhesion to vascular structures is one major reason for incomplete resection. After resection, sometimes long after resection, fusiform aneurysmal dilatation of adjacent arteries may be observed (Fig. 33-7). This has also been noted in other suprasellar tumors.17
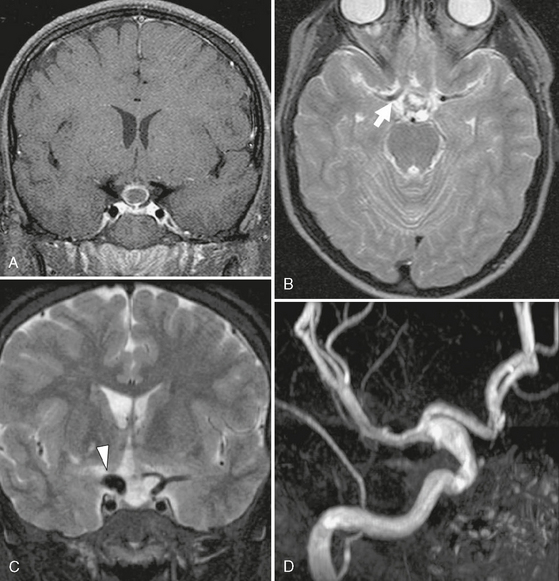
FIGURE 33-7 Craniopharyngioma. Coronal T1W postcontrast (A) and axial T2W (B) MR images demonstrate a small lesion in the suprasellar region that has rim enhancement. This was resected and after pathologic study was confirmed to be a craniopharyngioma. Note the normal size of the carotid terminus on the right (arrow, B). Postoperative coronal T2W MR image (C) and 3D TOF MRA (D) demonstrate a fusiform aneurysm involving the carotid terminus on the right (arrowhead, C).
METASTASIS, DIRECT TUMORAL SPREAD, AND PERINEURIAL SPREAD
Epidemiology
Hematogenous metastases to the pituitary and sellar region are uncommon, representing 0.14% to 28.1% of all brain metastases.18 They most frequently originate from lung and breast carcinomas. Direct local invasion most commonly results from squamous cell carcinoma, nasopharyngeal carcinoma, and rhabdomyosarcoma. Perineurial spread is most frequent with squamous cell carcinoma and adenoid cystic carcinoma of the head and neck.
Clinical Presentation
About 40% of patients with hematogenous metastases develop diabetes insipidus. Visual deficits and hypopituitarism are also common.18 Direct invasion by regional neoplasms usually causes symptoms from local mass effect. Perineurial spread may cause hypoesthesia and burning or stinging pain. Perineurial spread most frequently involves the fifth (trigeminal) and seventh (facial) cranial nerves, so it may be misdiagnosed as trigeminal neuralgia or Bell’s palsy. It may be asymptomatic in up to 45% of patients. There may be a long latency period between development of perineurial spread and tumor treatment, with reports of perineurial spread manifesting as late as 45 years after resection of the original tumor.20 Because perineurial spread changes the status of a lesion from resectable to nonresectable,19 and because many patients retain normal nerve function on clinical examination, imaging studies must try to identify perineurial spread that escapes clinical detection.
Pathophysiology
Hematogenous metastases to this region most frequently go to the pituitary gland, infundibulum, and tuber cinereum, because these structures lie outside the blood-brain barrier. Metastases to the infundibulum cause visual disturbance by compression of the chiasm or endocrine disturbance by disruption of the hypothalamic-pituitary axis. Hematogenous metastases to the pituitary gland itself most commonly pass to the infundibulum and posterior lobe, rather than the anterior lobe of the pituitary, because the posterior lobe is supplied directly through the inferior hypophysial branches of the internal carotid artery, whereas the anterior lobe is supplied indirectly via hypothalamic-portal vessels.18 The cavernous sinus may be involved by direct extension from regional neoplasms such as nasopharyngeal carcinoma, by hematogenous metastases, or by perineurial spread. Tumors may spread along any nerve, but perineurial spread most commonly involves the fifth and seventh cranial nerves. Squamous cell carcinoma and adenoid cystic carcinoma are the two most common tumors to spread by perineurial extension, but melanoma, lymphoma, leukemia, basal cell carcinoma, and mucoepidermoid carcinoma also reach the sellar-parasellar region by this route. Perineurial spread usually indicates a poor prognosis and may change the lesion’s status from resectable to nonresectable.
Imaging
CT
On CT, perineurial spread can be recognized by widening or destruction of the basal neural foramina, loss of the perineurial fat plane surrounding the nerves, and enhancement within the neural foramen after contrast agent administration. Growth of mass within the cavernous sinus bulges the lateral sinus wall outward. Normally, the lateral sinus wall is concave laterally. With a mass, it appears straightened or convex laterally. The internal carotid artery may be displaced, compressed, or occluded. There may be abnormal enhancement within the cavernous sinus.
MRI
Most pituitary metastases are difficult to distinguish from adenomas, especially if no other metastasis is identified. They typically appear as dumbbell-shaped intrasellar-suprasellar masses, indented at the level of the diaphragma sellae (Fig. 33-8). These lesions do not widen and remodel the sella because they grow too rapidly.21 Instead, they may destroy the sphenoid bone and invade into the cavernous sinus, partially occluding it. The flow void from the cavernous segment of the internal carotid artery may be displaced. Flow through it may be reduced or obstructed.
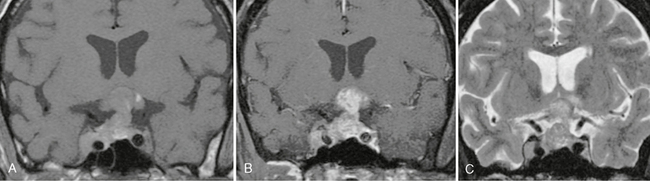
FIGURE 33-8 Renal cell metastasis. T1W MR image (A) demonstrates a lesion with intrinsic T1 shortening involving the sella and suprasellar regions, as well as extension into the right cavernous sinus. The lesion enhances avidly after administration of a contrast agent (B) and has slightly hyperintense signal on the T2W MR image (C).
Perineurial spread is identified by isointense thickening of the nerve, loss of the normal intraforaminal fat plane, and concentric enlargement of the foramen on T1W images. Contrast-enhanced studies show increased nerve size, nerve enhancement, and widening of the cavernous sinus (Fig. 33-9). Fat suppression can help to identify the abnormal enhancement. Atrophy of the muscles supplied by the affected nerves is an important secondary sign of perineurial invasion. Because nasopharyngeal carcinoma is a common source of perineurial spread, imaging of the parasellar region should always include the nasopharynx.
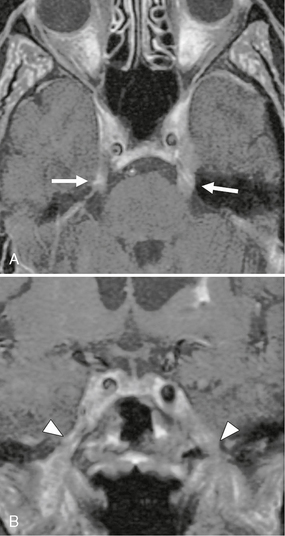
FIGURE 33-9 Lymphoma with perineurial spread. A, Axial T1W postcontrast MR image demonstrates thickening and increased enhancement along the bilateral cisternal segment of the trigeminal nerve (arrows), and there is a prominence of the cavernous sinus. B, Abnormal enhancement and thickening of V3 extending through the foramen ovale is noted on this coronal image (arrowheads).
LANGERHANS CELL HISTIOCYTOSIS
Epidemiology
Langerhans cell histiocytosis is a rare disorder, seen in up to 2 per 100,000 people.22 It most frequently affects children and adolescents but can occur in adults. There is no gender predilection.
Pathophysiology
Langerhans cells are a dendritic cell line derived from bone marrow cells. The cells have antigen-presenting and antigen-processing properties22 and are normally found in the pituitary and brain parenchyma. The pathophysiologic mechanism of Langerhans cell histiocytosis has not been elucidated.