• The optic canal and SOF open into the orbit. Just caudal to the SOF, the FR lies in a posterior-anterior orientation through the skull base. The FR passes from the CS to the pterygopalatine fossa (PPF) (Fig. 3-2), entering the fossa at the level of the inferior orbital fissure (IOF). The V2 (maxillary) division of the trigeminal nerve, carried through the FR, crosses the upper part of the fossa and enters the orbit through the IOF. It becomes the infraorbital nerve in the floor of the orbit and exits through the infraorbital foramen to the face.
• The pterygoid canal (PC, vidian canal) is inferomedial to the FR, transmitting the pterygoid nerve and artery from the anterior wall of foramen lacerum to the PPF.
• The foramen ovale (FO) is anterior and medial to the foramen spinosum (FS) (Fig. 3-3). The FO transmits the V3 (mandibular) division of the trigeminal nerve into the masticator space, as well as the accessory meningeal artery and the lesser superficial petrosal nerve. The FS transmits the middle meningeal artery and the recurrent branch of the mandibular nerve.
• The PPF is an important landmark that provides pathways for tumor spread by its connections: via the PC and FR to the central skull base and middle cranial fossa; via the IOF to the orbit; via the pterygomaxillary fissure to the infratemporal fossa; via the sphenopalatine foramen to the nasal cavity; and via the pterygopalatine canals to the oral cavity.
• Posterior cranial fossa, containing the cerebellum and brain stem, is formed by the dorsum sellae and clivus of the sphenoid, the occipital bone, the petrous and mastoid temporal bone, and the mastoid angle of the parietal bone. Cranial nerves VII–XII exit in this fossa.
• The jugular foramen is situated between the lateral occipital bone and the petrous temporal bone (Fig. 3-1). Above the jugular foramen is the internal acoustic meatus, which transmits the facial and acoustic nerves and the internal auditory artery.
3. NATURAL HISTORY
3.1. Meningioma
• Meningiomas represent approximately 15% of primary brain tumors and occur with an annual incidence ranging from <1 to >6 per 100,000.1,2
• They arise from the arachnoidal cells of the meninges.3 The peak incidence is during the sixth and seventh decades of life.
• The ratio of males to females ranges from 1:1.4 to 1:2.8.1,4,5
• Most meningiomas occur along either the convexities or the parasagittal plane. Six morphologic variants defined by the World Health Organization (grade II: atypical, clear cell, chordoid; grade III: anaplastic, papillary, rhabdoid) are associated with aggressive clinical behavior and increased recurrence and metastasis.6
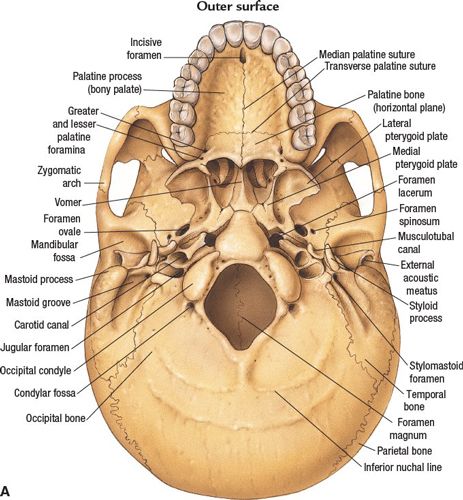
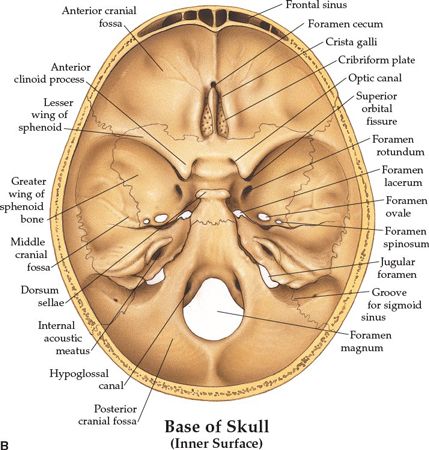
FIGURE 3-1. Base of skull. (A) Outer surface. (B) Inner surface. (From Anatomical Chart Company copyright © 2008, Lippincott Williams & Wilkins. All rights reserved.)
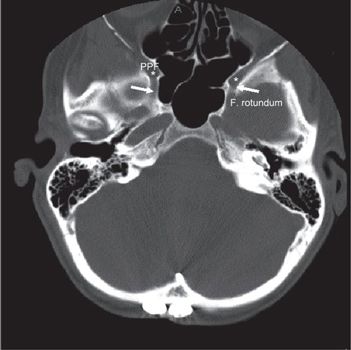
FIGURE 3-2. Foramen rotundum (white arrow) and pterygo-palatine fossa (star, PPF) demonstrated in axial CT slide with head and neck bone image resolution.
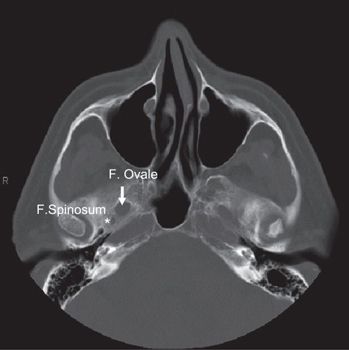
FIGURE 3-3. Foramen ovale (arrow) and foramen spinosum (star) demonstrated in axial CT with head and neck bone image resolution.
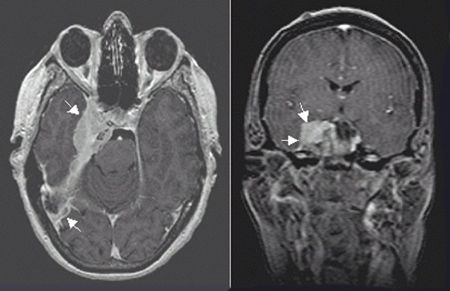
FIGURE 3-4. Right-sided petroclival meningioma with a characteristic dural-based tail appearance and parasellar and cavernous extension in axial (left) and coronal (right) T1 MRI with gadolinium.
• Although skull base origin is infrequent, they may arise from any part of the sphenoid such as the greater wing, the planum sphenoidale, the tuberculum sellae, the walls of the CS, petrous apex or clivus (petroclival), parasellar regions (Fig. 3-4), the optic sheath, the petroclinoid, or the trigeminal nerve.
• Skull base meningiomas extend along the dural surfaces or intraosseously. Penetrating through bone may cause a substantial extracranial mass.
• As arachnoid cells accompany the cranial nerves, meningiomas can be found adjacent to and traversing various neural foramina.7
3.2. Esthesioneuroblastoma
• Esthesioneuroblastoma (olfactory neuroblastoma, neuroendocrine carcinoma) constitutes 1% to 5% of all malignant neoplasms of the nasal cavity.8;9 It originates from the basal cells of the olfactory epithelium in the upper third of the nasal septum, the cribriform plate, or the superior turbinates (Fig. 3-5). It has a spectrum of neural and epithelial differentiation.10,11
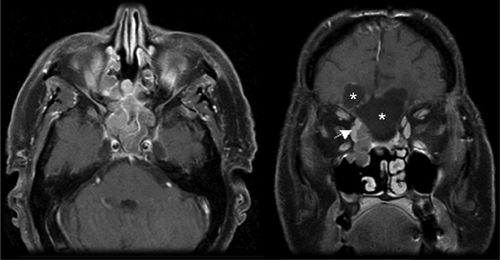
FIGURE 3-5. Postsurgical resection of right-sided esthesioneuroblastoma with a residual right medial orbital mass (white arrow) in axial (left) and coronal (right) T1 MRI with gadolinium. Postoperative cystic areas are marked (stars) in coronal section.
• It has a fairly equal distribution between sexes, with a slight male predominance. The mean age at diagnosis is 45 years, with a bimodal distribution with peaks of incidence in the second and fifth decades of life.12
• Early symptoms are usually nasal obstruction or epistaxis. Periorbital swelling and hyposmia are also common.
• Advanced age, brain involvement, high histologic grade, and advanced Kadish stage (Table 3-4) are major prognostic factors. Polin et al. reported that, in their experience at the University of Virginia, advanced Kadish stage was associated with a slightly higher rate of disease-related mortality, but not with disease-free survival.13
• Synchronous cervical lymph node metastases incidence is 17% to 47%.14
• Distant hematogenous metastases are not usual at presentation, but may occur after relapse. Metastases occur mainly to the lung, bone, bone marrow, or skin.9
3.3. Chordoma
• Chordomas represent approximately 1% of all malignant bone tumors and arise from remnants of the notochord. The terminus of the notochord is in the sphenoid bone, so skull base chordomas arise in the region of the clivus (Fig. 3-6).
• Thirty-five percent of chordomas occur in the skull base, 50% in the sacrococcygeal region, and 15% in the spine.15
• Most chordomas in the craniovertebral area are diagnosed in persons aged 30–50 years, while sacrococcygeal chordomas have their peak incidence at 40–60 years of age.
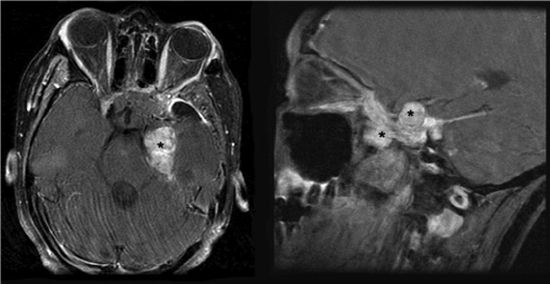
FIGURE 3-6. Left-sided chordoma tumor (black stars) with compression of brain stem in axial (left) and coronal (right) T1 MRI with gadolinium.
• Male predominance has been reported,15 while in another series, the gender distribution was even.16
• The three pathologic subtypes are described as classic (most common), chondroid (mostly in the skull base), and dedifferentiated (rare, but poor prognosis).15,17
• Distant metastases, more common in sacral and vertebral chordomas than in skull base chordomas, occur in the lungs, bones, lymph nodes, liver, and skin.18–20 Intradural metastases may appear following surgical resection of skull base chordomas, but are rare.21
3.4. Glomus Jugulare Tumor
• Glomus jugulare tumor (GJT)—also known as paraganglioma or chemodectoma—is a rare tumor with an incidence of about 1 per 1,000,000 population.22
• It is the most common neoplasm of the middle ear.23,24 It is the most commonly diagnosed neurotologic neoplasm after acoustic neuroma, and originates from special neural crest elements called paraganglion cells, which, along with autonomic ganglion cells, form the paraganglia. It is composed of two types of cells: chief cells and sustentacular cells (modified Schwann cells).
• It is part of the diffuse neuroendocrine system. The chief cells of the glomus body are nearly identical to the chromaffin cells of the sympathetic ganglia and contain osmiophilic granules similar to those implicated in catecholamine storage.25,26 Glomus bodies may function in a manner similar to that of the carotid body as chemoreceptors, responding to alterations in blood pH and oxygen and carbon dioxide tensions, as well as affecting systemic blood pressure through the release of various transmitters.
• Glomus bodies have a rich blood supply and are innervated by either the tympanic branch of the glossopharyngeal nerve (Jacobsen’s nerve) or the auricular branch of the vagus nerve (Arnold’s nerve).25
• Although usually benign and slow growing, they are destructive. In about 3% of patients, the tumor is malignant.25
• A very strong female predominance of 4:1 to 6:1 has been reported.27,28
• Typically, these tumors develop during the fifth and sixth decades of life.27
• GJTs have their epicenter in the region of the jugular bulb. Neurovascular structures within the hypoglossal canal, jugular foramen, and temporal bone can be affected (Fig. 3-7).
• Generally, patients with a GJT exhibit tinnitus; cephalagia; hyperacusis; anacusis; dizziness; or multiple injuries of cranial nerves VII, VIII, IX, X, XI, and XII.
3.5. Medulloblastoma
• Medulloblastoma represents 20% of childhood brain tumors. The median age at diagnosis of childhood medulloblastoma is 6 years, and approximately 25% occur in children younger than 3 years. Medulloblastoma in adults occurs predominantly in the third to fifth decades of life and constitutes 10% to 20% of medulloblastomas.
• Headaches, vomiting (a sign of increased intracranial pressure), and ataxia are the classic presenting triad of posterior fossa tumors.
• Most often arising in the cerebellar vermis (Fig. 3-8), the tumor grows to fill the fourth ventricle and frequently attaches to the brain stem. It has a characteristic seeding tendency associated with subarachnoid dissemination through the cerebrospinal fluid (CSF). Subarachnoid disease is detected in 20% to 25% of cases at diagnosis.29,30
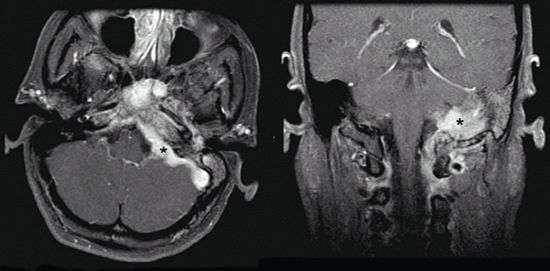
FIGURE 3-7. Left-sided glomus jugulare tumor (black stars) with extension to mastoid air cells, infratemporal fossa, cerebellopontine angle, and cavernous sinus in axial (left) and coronal (right) T1 MRI with gadolinium.
3.6. Glial Tumors
• Glial tumors are primary central nervous system neoplasms of neuroglial cells such as astrocytes or oligodendrocytes demonstrating various characteristics as low-grade gliomas (LGG) with slower growth pattern and high-grade gliomas (HGG) with rapid progression.
3.6.1. Low-Grade Glial Tumors
• LGG account for approximately 10% of primary CNS tumors.31 Histologic subtypes of LGG include pilocytic astrocytomas (PA) classified as WHO Grade I, diffuse infiltrating LGG such as oligodendrogliomas, and mixed oligoastrocytomas classified as WHO Grade II.32 Grade II defines well-differentiated gliomas without demonstration of mitoses, nuclear pleomorphism, anaplasia, vascular proliferation, and necrosis.
3.6.2. High-Grade Glial Tumors
• Glioblastoma (GB) and anaplastic gliomas account for approximately one-third of primary brain tumors.31 GB is the only WHO grade IV glioma that is astrocytoma with nuclear atypia, mitotic activity, vascular proliferation, and necrosis.32 Anaplastic gliomas classified as WHO grade III tumors include anaplastic astrocytoma (AA), anaplastic oligoastrocytoma (AOA), anaplastic oligodendroglioma (AOD), and anaplastic ependymoma.32 AA exhibits mitoses but no endothelial proliferation or necrosis. AOD is an oligodendroglial tumor with focal or diffuse histologic malignancy features and a less favorable prognosis than that of grade II oligodendroglioma.
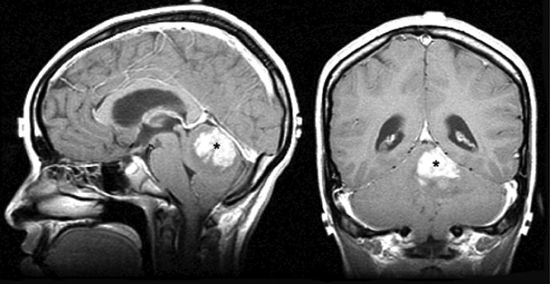
FIGURE 3-8. Medulloblastoma of the cerebellar vermis (black stars) in sagittal (left) and coronal (right) T1 MRI with gadolinium.
• All high-grade gliomas express vascular endothelial growth factor (VEGF) as well as oligodendrocyte lineage transcription factor 2 (OLIG2); besides, there have been genetic alterations leading to primary GB, secondary GB, and AOA.33 MGMT promoter methylation and IDH1 mutational status allow for stratification of glioblastoma patients into prognostically distinct subgroups.34
4. DIAGNOSIS
4.1. Signs and Symptoms
• Common presenting symptoms of skull base tumors are orbitofrontal headaches due to involvement or stretching of the dura and visual disturbances due to involvement of optic nerves or neural contents of the cavernous sinuses.
• Frontal base and paranasal extension may cause olfactory and frontal syndromes, such as increased intracranial pressure, seizures, or personality changes for subfrontal tumors and loss of sensation of smell, nasal obstruction, rhinorrhea, or epistaxis for paranasal involvement.
• Tumors affecting the sella turcica and sphenoid sinus cause chiasmatic or hypothalamic symptoms. Chiasmatic involvement results in vision loss due to hemianopsia, either unilateral or bitemporal. Endocrinologic symptoms such as diabetes insipidus, amenorrhea, impotence, or pituitary apoplexy can be observed in cases of pituitary or hypothalamic involvement.
• Tumors involving the lateral middle fossa, infratemporal fossa, or sphenoid wing may extend into the orbit and induce exophthalmus, diplopia, or unilateral vision loss.
• Medial middle fossa tumors, including those of the CS and MC, may present as dysfunction of one or more of the upper cranial nerves (III, IV, V1–3, VI). Tumors in this area may encase or constrict the carotid artery in the parasellar region causing cerebral ischemia.
• Petroclival–clival involvement may result in cranial nerve deficits, cerebellar signs, corticospinal tract involvement, or increased intracranial pressure.
• Internal auditory canal or cerebellopontine angle involvement may cause hearing loss, or deficits secondary to cerebellar or brainstem compression.
• Different lobes involved with high-grade glial tumors may initiate various signs and symptoms such as seizures, hemiparesis, mental status lability, urinary incontinence for the frontal lobe; seizures, language/reading/calculation disturbances for the parietal lobe; seizures, aphasia, emotional disturbances for the temporal lobe; seizures and visual changes for the occipital lobe; etc.
4.2. Physical Examination
• Evaluation requires clinical examination for cranial neuropathy and imaging evidence for extent of disease in patients known to have a skull base lesion. Physical examination is based mainly on the general neurologic examination, including cranial nerves for signs of skull base lesions and basic cerebellar functions for signs of posterior fossa tumors.
4.3. Imaging
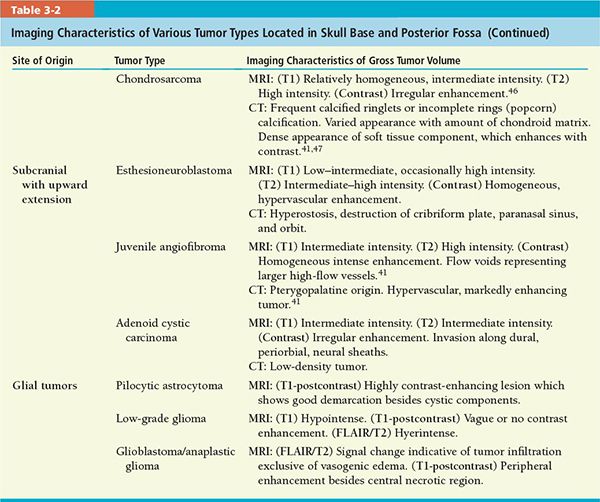
• Neuroimaging is critical for pretreatment planning and posttreatment surveillance.35 Detection of differential features might predict tumor histology and help in delineation. Important features include calcification, mineralized matrix, hyperostosis, enhancement pattern, flow voids on magnetic resonance imaging (MRI), and hypervascularity on computed tomography (CT).
• CT scan and MRI are, in most cases, complementary in diagnosis and treatment of skull base tumors.
• CT scan is superior for detecting calcification in the tumor as well as slight cortical erosion or frank bony destruction.
• MRI is better for detecting soft tissue details such as intracranial extension, compression, involvement of the neural foramina, perineural tumor spread, vascular encasement, and thrombosis.
• A slow-growing or more benign process usually presents with remodeled or pushed away bony structures. A more aggressive lesion should be favored if there is gross destruction of bone (chewed up, moth-eaten).
• Most tumors affecting bone replace healthy bone marrow (T1 hyperintense on MRI) with isointense tumor.
• Bony hyperostosis associated with a soft-tissue tumor usually suggests meningioma, and occasionally more malignant tumors such as esthesioneuroblastoma. Hyperostosis without a soft tissue component may be fibrous dysplasia, Paget’s disease, or osteoblastic metastasis (e.g., prostate cancer).
• Clinical correlation is required to identify extension, such as denervation (masticator musculature problems: MC or FO; ipsilateral tongue paralysis: hypoglossal canal).
• Clival tumors with gross destruction, bony fragments, and bright signal on T2-weighted MRI suggest chordoma.
• A jugular foramen origin with a moth-eaten bony pattern, with or without cranial neuropathy or tinnitus, suggests a GJT.
• Nondestructive enlargement of a neural foramen or transforaminal tumor extension suggests schwannoma or meningioma.
• An enhancing tumor in the internal auditory canal, presenting with sensorineural hearing loss (SNHL), very strongly suggests acoustic schwannoma.
• Tumor involvement of the sella, parasellar region, and/or suprasellar cistern, with central skull base destruction, should prompt consideration of invasive pituitary adenoma.
• A review of imaging characteristics of selected tumors is given in Table 3-2.

4.4. Staging
• Meningioma: Intracranial meningiomas are generally staged according to the extent of surgical resection (Table 3-3).48
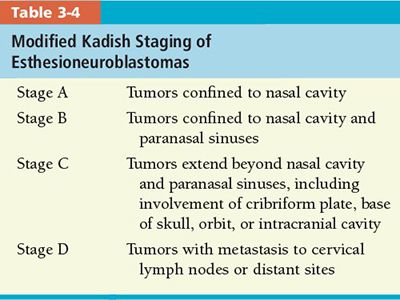
• Esthesioneuroblastoma: Kadish et al.49 developed a staging system, which has been modified by Morita et al.,12 based on anatomic extension of the tumor beyond the nasal cavity (Table 3-4).
• Chordoma: Various classification schemes based on location have been proposed: clival, parasellar, and sellar;50 basiocciput-caudal and basisphenoid-rostral;51 and, of greatest help in the choice of surgical approach, superior, middle, and inferior clival.52
• Glomus Jugulare Tumor: Several classification systems have been proposed over the years. The most recent and widely used systems are the Fisch classification53 (Table 3-5), revised in 1981, and the Glasscock and Jackson classification, based on site and extent of the tumor.54
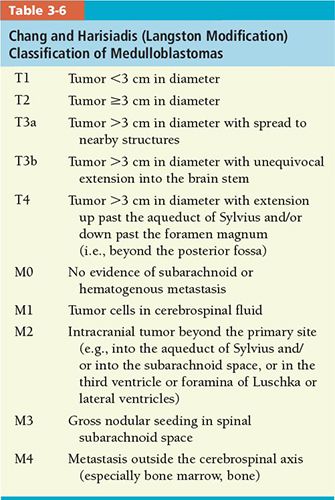
• Medulloblastoma: The most widely used classification is the Chang operative staging system (Table 3-6).55 The degree of residual tumor and presence or absence of subarachnoid or extraneural metastases determine stage.
• Glial tumors: The WHO grading establishes a malignancy scale based on histologic features (Table 3-7)32,56. The low grade constitutes grades I and II, and high grade, III and IV.


5. GENERAL MANAGEMENT
5.1. Meningioma
• The treatment of choice is complete surgical excision, but this is not always possible, as 20% to 50% of meningiomas are located in the skull base and may not be amenable to a safe surgical approach.57

• Surgical literature reports permanent cranial nerve deficits in 22% to 91% of patients undergoing surgical resection for petroclival meningiomas; an average of 54% develop new onset of neurologic symptoms after surgery.58–62
• To reduce the risk of neurologic deficit, a subtotal resection is often accepted. However, adjuvant therapy is usually required in these cases, since an estimated 10% to 56% of patients develop recurrence after subtotal resection.63–65 In a number of series, fractionated radiotherapy significantly increased tumor control rates after subtotal resection of meningiomas.66–69
• Goldsmith et al. found 5-year progression-free survival rates of 87% for benign and 45% for malignant meningiomas treated with radiation after subtotal resection, and a 5-year progression-free survival rate of 98% in a subset of patients with benign lesions treated in the era of CT-/MRI-based planning.67
• Smaller series looking specifically at malignant or atypical meningiomas, including data from our own institution, have also demonstrated a benefit from radiation.70,71
• Similarly, stereotactic radiosurgery has been used in the treatment of meningiomas. Recently, Kondziolka et al. reported a 93% clinical tumor control rate in a series of 99 patients with a minimum of 5 years of follow-up, and recommended gamma knife treatment for lesions <3 cm in diameter with regular borders that do not compress the optic chiasm.72
• Skull base meningiomas that appear progressive in serial imaging or are symptomatic deserve treatment. Surgery is the first step if operable with minimal toxicity. Adjuvant radiotherapy (3DCRT, IMRT, or stereotactic radiosurgery) is indicated for WHO grade-II and -III meningiomas, even with gross total resection (GTR), and for WHO grade I tumors after subtotal resection (STR) if future salvage gross total resection seems not possible.
• Radiation therapy can be delivered as 3DCRT, IMRT, SRS, FSRT, or proton-beam therapy.
• Recommended dose is 50 to 54 Gy in 25–30 fractions for grade I [clinical target volume (CTV) = contrast-enhancing gross tumor volume (GTV) in T1 weighted MRI), and 57 to 60 Gy in 30–33 fractions for grade II to III tumors. Recommended SRS dose (up to 3 to 4 cm) is 12 Gy in 1 fraction when the tumor is adjacent to critical structures such as the brainstem or optic nerves, and 12 to 18 Gy (higher for grades II to III) for others clear from critical structures (18 Gy if <1 cm, 16 Gy if 1 to 3 cm, 12 to 14 Gy if >3 cm) (Fig. 3-9).73–79
• The European Organisation for Research and Treatment of Cancer (EORTC) has launched the phase II trial 22042-26042,80 focusing only on WHO grade II and III tumors, which examines the utility of adjuvant postoperative high-dose fractionated radiotherapy. Patients with WHO grade II tumors are stratified into two groups based upon the Simpson grade (SG) of resection; SG 1–3 are treated with fractionated radiotherapy to 60 Gy, and SG 4 or 5 are treated with 60 Gy plus a boost of 10 Gy. Patients with WHO grade III tumors are also similarly separated into two groups for a descriptive observational study.
• The Radiation Therapy Oncology Group (RTOG) has launched phase II trial 053981 for all grades of meningioma through three groups based on the likelihood of tumor regrowth after surgery risk. Group I tumors (WHO grade I after any degree of surgical resection) are observed postoperatively without adjuvant therapy until recurrence. Group II tumors (recurrent grade I or newly diagnosed grade II following SG 1–3) are treated with conformal or intensity modulated radiotherapy (IMRT) to 54 Gy in 30 fractions. Group III tumors (any grade III, any recurrent grade II, or any newly diagnosed WHO grade II following SG 4 or 5) are treated with IMRT to 60 Gy in 30 fractions.
5.2. Esthesioneuroblastoma
• Preferred treatment in localized tumors is en bloc craniofacial resection of the tumor, cribriform plate, and overlying dura.82
• In advanced disease, preoperative radiotherapy with or without chemotherapy is preferred. An initially planned combination of surgery followed by postoperative radiotherapy may be another option.
• Cyclophosphamide, vincristine, and doxorubicin are generally preferred for concomitant radiotherapy. Platinum-based chemotherapy is preferred for advanced, high-grade tumors.
• Primary radiotherapy is preferred only in selected patients who are ineligible for surgical resection.
• Positive surgical margins are the most significant predictor of progression and decreased overall survival rates in the experience of many centers.83–86
• Radical surgery alone is not enough to avoid local recurrences; adjuvant postoperative high-dose radiotherapy has achieved high local control rates (92%) in a recently reported series.86–88
• Nearly one-half of the recurrences occur >5 years after diagnosis.89 Recurrence rates range from 30% to 70%.
• Estimated survival rates are 74% to 87% at 5 years and 54% to 60% at 10 years. Five-year survival rate following local recurrence is 82% after salvage therapy.
• We recommend as radical a surgical procedure as possible with preoperative or postoperative radiotherapy (dose ~60 Gy) and concomitant chemotherapy, as aggressive, multimodality treatment is supported by the findings of many centers.13,86,90,91
• The efficacy of IMRT on tumors of nasal cavity and paranasal sinuses are more commonly reported in recent series.92–102
5.3. Chordoma
• Wide or complete resection is the treatment of choice, but is not feasible in most cases. Adjuvant radiation has been advocated for patients with positive surgical margins and residual disease.103–105
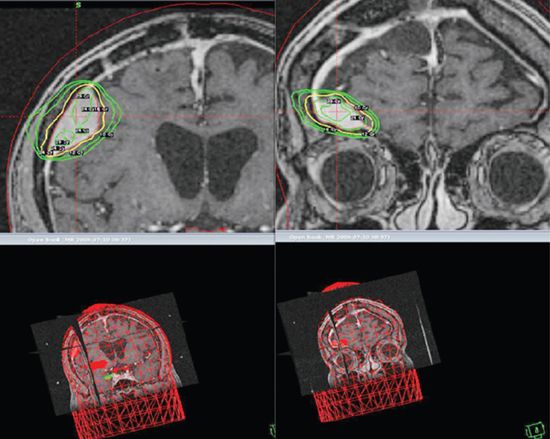
FIGURE 3-9. Two separate right posterior and anterior frontal meningiomas radiosurgically treated with a single fraction of 16 Gy.
• Munzenrider and Liebsch suggested that charged particle beams are ideal for treating skull base chordomas because such beams can deliver precise, high-dose radiation to localized areas of disease while sparing surrounding normal structures.106 Proton–photon therapy was recommended by both Borba et al. and Habrand et al. for intracranial tumors in children.107,108 The 5-year actuarial survival rate was reported as 68%, while the disease-free survival rate was 63% for intracranial chordomas.
• Romero et al. and Klekamp et al. produced comparable results with high-dose fractionated conventional radiotherapy.109,110 Romero et al. showed that prognosis for patients with microscopic residual disease given 48 Gy postoperative radiotherapy was better than for those who received <40 Gy. Klekamp and colleagues reported that postoperative radiation doses of 60 to 70 Gy assured a significantly longer recurrence-free interval in contrast to suboptimal excision without radiotherapy.
• IMRT, which can deliver precise, high-dose radiation while sparing surrounding normal structures, has not yet been reported to emphasize the comparison in results.
• The use of charged particle radiotherapy with protons as the postoperative treatment in single-institutional retrospective studies suggests better results as compared to the use of conventional photon irradiation; the former yields better 10-year outcome with relatively few significant complications despite the higher doses delivered with this therapeutic modality.111,112
• We prefer, if possible, to prescribe 66 to 70 Gy in 2 Gy/fraction given by IMRT, FSRT, or proton radiotherapy (Fig. 3-10).
5.4. Glomus Jugulare Tumor
• Management of jugular paraganglioma historically has shown a pendulum swing away from virtually exclusive primary surgery to more of primary radiation therapy, with a very recent moderating trend toward selecting treatment according to patient profile.
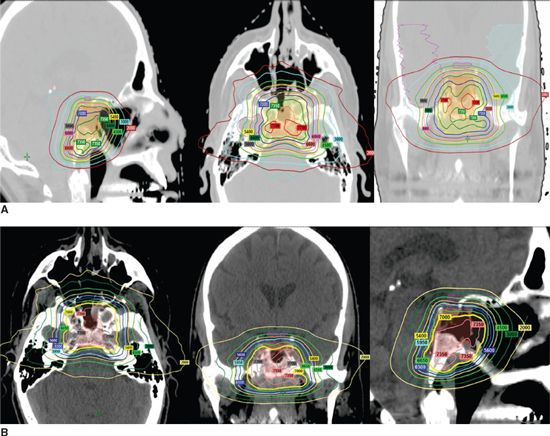
FIGURE 3-10. Proton radiotherapy for clivus (A) and parasellar (B) chordoma cases delivered 70 Gy while sparing brainstem with a sharp dose decrease.
• The goal of surgery is complete tumor removal, whereas the goal of conventionally fractionated, external beam radiation therapy and stereotactic radiosurgery is long-term tumor control by preventing tumor growth and regional extension that could lead to progressive symptoms and neurologic deficits.
• Local tumor control rates after total surgical removal vary from 0% to 90%.113 Surgical removal can be associated with considerable morbidity and mortality.114–118 In general, gross total surgical resection has been accomplished in 40% to 80% of cases in different series.119–121
• Progressive growth of GJTs was arrested by conventional external beam radiation therapy at rates of local control ranging from 85% to 100%, with reversal of symptoms in some patients, and complication rates of 0% to 10%.122–125
• Despite the inherent limitations and biases of retrospective, predominantly single-institution reported series, surgery and radiation therapy appear to have similar rates of long-term local control (~90%). This is true even though patients treated with radiation therapy generally have larger or more infiltrative tumors that are not readily subject to surgical resection. Disease-related mortality is only limited to a few patients with residual or recurrent tumor, irrespective of the mode of treatment. Therefore, treatment of GJTs should focus primarily on decreasing morbidity.
• A primary surgical approach is recommended for younger patients with lesions that are readily resectable without significant neurologic impairment. This approach avoids the small risk of radiation-induced second malignancy.
• Involvement of the internal carotid artery (demonstrated by balloon occlusion test), contralateral SNHL, bilateral tumors with contralateral deficits of the lower cranial nerves, concerns of venous return, and poor medical condition are common causes for inoperability and therefore are indications for primary definitive radiotherapy.
• We recommend a conventionally fractionated IMRT approach for most patients whose tumors are irregularly shaped and are in close proximity to radiosensitive normal structures. A dose of 45 to 50 Gy (1.8 to 2 Gy/fraction) is prescribed, encompassing radiographically visible tumor as CTV plus a 3-mm margin for setup uncertainty, making up the planning target volume (PTV).
5.5. Medulloblastoma
• The initial goal is maximal surgical resection for both treatment and reestablishing CSF flow. Gross total resection (>90% of the tumor removed, with <1.5 cm2 residual) generally is limited only by substantial invasion of the tumor into the brain stem or infiltration into the peduncle. Gross or near total resection is achieved in 90% of children with medulloblastoma older than 3 years in the USA.126
• MRI brain imaging within the immediate postoperative period (1 to 3 days) is important to define postoperative residual effect, which is a reliable prognostic factor.126
• Neuraxis staging is recommended 10 days or longer after surgery to eliminate the confusion between findings and postoperative debris. A contrast-enhanced spinal MRI and subsequent lumbar CSF cytologic studies are standard.
• Categorization into standard (or average-risk) and poor-risk disease guides the treatment decision. Average-risk is defined as patients older than 3 years with posterior fossa tumors who underwent total or “near-total” (<1.5 cm2 of residual disease) resection with no dissemination.55,127
• Postoperative management currently consists of radiotherapy and chemotherapy.128
• Radiotherapy requires systematic inclusion of the entire subarachnoid space-craniospinal irradiation (CSI) followed by a boost to the posterior cranial fossa. The conventional dose to the neuraxis has historically been 36 Gy (1.8 Gy/day). Gross total resection and postoperative CSI yield a 5- to 10-year progression-free survival rate of approximately 65%.129–133
• Pediatric Oncology Group and Children’s Cancer Group conducted a randomized study that compared CSI at doses of 36 and 23.4 Gy in average-risk medulloblastoma, and established a standard of removing >90% tumor with less than 1.5 cm2 of residual disease.133 Reduced-dose neuraxis irradiation (23.4 Gy) was associated with higher risk of early relapse and early isolated neuraxis relapse, and lower 5-year rates of event-free survival96
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


