Chapter 37 Soft Tissue Sarcomas
Introduction
Soft tissue sarcomas are rare mesenchymal tumors that originate from the mesoderm, with the notable exceptions of those arising from primitive neuroectodermal tissue and those with unknown cell derivation, such as Ewing’s sarcoma or synovial sarcoma.1,2 Because more than 50 clinically and molecularly distinct sarcoma subtypes exist, there is tremendous clinical variation that leads to marked heterogeneity in their respective clinical behavior, prognosis, metastatic risk, and chemotherapy responsiveness.3 For this reason, although many soft tissue sarcomas are treated similarly, clinicians must be aware of the subtype-dependent nuances. Despite having nearly the same incidence as multiple myeloma or thyroid carcinoma, soft tissue sarcomas are responsible for more deaths than testicular tumors, Hodgkin’s disease, and thyroid cancer combined.4,5 They are two to three times more common than primary malignant bone tumors, and despite apparently complete surgical resection, they often metastasize to the lungs.
Epidemiology and Etiologic Considerations
Soft tissue sarcomas account for approximately 1% of all primary adult malignancies and 7% to 15% of pediatric neoplasms.2,6 The general incidence of these tumors is 1.4 per 100,000, but it rises to 8 per 100,000 in people older than 80 years. Roughly 40% of all soft tissue sarcomas occur in people older than 50 years.2 The American Cancer Society estimates a combined incidence of soft tissue sarcomas (including heart) in adults and children of about 10,980 new cases (6050 males and 4930 females) and about 3920 deaths (2060 males and 1860 females) for the year 2011.7 The overall incidence has been gradually increasing, in part due to better diagnosis but also probably due to a true increase in acquired immunodeficiency syndrome (AIDS)–associated Kaposi’s sarcoma.8 A male-to-female ratio of approximately 1.2 to 1 has been reported, although this varies considerably depending upon the sarcoma subtype.2 The relative frequency of each subtype of soft tissue sarcoma varies according to age. For instance, rhabdomyosarcoma mostly occurs in children, synovial sarcoma in young adults, and malignant fibrohistiocytoma (MFH) in older populations. Benign soft tissue tumors are approximately 100 times more common than malignant soft tissue tumors. Soft tissue sarcomas are most often seen in the extremities (59%), trunk (19%), retroperitoneum (15%), and head and neck (9%).2
The vast majority of soft tissue malignancies occur sporadically without any predisposing cause. However, soft tissue sarcomas may occur 3 to 15 years after irradiation for lymphoma, cervical cancer, breast cancer, or testicular cancer.9 Viruses have also been implicated with some soft tissue sarcomas, such as leiomyosarcoma, and with Kaposi’s sarcoma in patients with AIDS.10 Chronic lymphedema-associated angiosarcoma (Stewart-Treves syndrome) occurs as a rare complication of breast cancer treatment.11 An increased incidence of soft tissue sarcomas has been reported in several genetic disorders, such as neurofibromatosis, hereditary retinoblastoma, and Li-Fraumeni syndrome.12 Chemicals, such as Agent Orange (a dioxin-containing herbicide) and immunosuppressive drugs, are an uncommon cause of soft tissue sarcoma.13
Key Points Epidemiology and risk factors
• Soft tissue sarcomas can be seen at any age; most occur after age 50 years.
• They make up approximately 1% of all primary tumors in adults and 7% to 15% in children.
• They most commonly occur in the lower extremities.
• Most soft tissue sarcomas occur without any predisposing cause. In a few instances, genetic, infections (viral), chemicals (Agent Orange), physical agents (radiation), and immunosuppressive drugs may be contributory.
Histopathologic Considerations
The World Health Organization (WHO) classifies more than 50 histologic subtypes of soft tissue sarcomas as shown in Table 37-1.3 Thus, histopathologic diagnosis of soft tissue sarcoma is essential before treatment. In general, biopsy should be performed on any symptomatic or enlarging soft tissue mass that persists for longer than 4 weeks or is larger than 5 cm in diameter.5
Table 37-1 Modified World Health Organization Classification of Soft Tissue Sarcomas
| TUMOR TYPE | TUMOR |
|---|---|
| Adipocytic | |
| Intermediate (locally aggressive) | Atypical lipoma (well-differentiated liposarcoma) |
| Malignant | Liposarcoma: dedifferentiated, myxoid, round cell, pleomorphic, mixed type, not otherwise specified |
| Fibroblastic/Myofibroblastic | |
| Intermediate | |
| Locally aggressive | Superficial fibromatosis, desmoid-type fibromatoses, lipofibromatosis |
| Rarely metastasizing | Solitary fibrous tumor and hemangiopericytoma, infantile fibrosarcoma |
| Malignant | Adult fibrosarcoma, myxofibrosarcoma |
| So-called Fibrohistiocytic | |
| Intermediate (rarely metastasizing) | GCT of soft tissues |
| Malignant | Pleomorphic fibrous histiocytoma or undifferentiated pleomorphic sarcoma (MFH), giant cell fibrous histiocytoma or undifferentiated pleomorphic sarcoma with giant cells, inflammatory fibrous histiocytoma or undifferentiated pleomorphic sarcoma with prominent inflammation |
| Smooth Muscle | |
| Malignant | Leiomyosarcoma |
| Pericystic (Perivascular) | Malignant glomus tumors, malignant hemangiopericytoma |
| Skeletal Muscle | |
| Malignant | Rhabdomyosarcoma: embryonal, alveolar, pleomorphic, botyroid, spindle cell, and rhabdomyosarcoma with ganglionic dedifferentiation |
| Vascular | |
| Intermediate | |
| Locally aggressive | Kaposiform hemangioendothelioma |
| Rarely metastasizing | Retiform hemangioendothelioma, Kaposi’s sarcoma |
| Malignant | Epithelioid hemangioendothelioma, angiosarcoma of soft tissue |
| Chondro-osseous | Mesenchymal chondrosarcoma, extraskeletal osteosarcoma |
| Uncertain Differentiation | |
| Intermediate (rarely metastasizing) | Angiomatoid fibrous histiocytoma, ossifying fibromyxoid tumor |
| Malignant | Synovial sarcoma, epithelioid sarcoma, clear cell sarcoma of soft tissue, extraskeletal myxoid chondrosarcoma, extraskeletal Ewing’s tumor, intimal sarcoma, extraskeletal osteosarcoma, alveola soft part sarcoma |
| Neurogenic | |
| Malignant (peripheral nerve sheath) | Neurofibrosarcoma, malignant schwannoma |
| Pleuripotential Mesenchymal Tissue | Malignant mesenchymoma |
GCT, germ cell tumor; MFH, malignant fibrous histiocytoma.
Computed tomography (CT)– or ultrasound (US)–guided percutaneous fine-needle aspiration (FNA) biopsy is performed under local anesthesia. An FNA biopsy has low risk of complications but a greater probability of misdiagnosis, and its limitations make it best for suspected recurrent soft tissue tumors or nodal metastases.5,14
A core needle biopsy is often preferred because it has relatively low (~1%) rate of complications, and a much higher diagnostic yield than an FNA biopsy.14–16 Several specimens should be obtained within a soft tissue tumor to ensure adequate tumor tissue for various pathologic stains, electron microscopic studies, cytogenetic analysis and flow cytometric studies. US, CT or gadolinium-enhanced magnetic resonance imaging (MRI) is often used as a guide to determine the best area for biopsy. In addition, the biopsy site should be chosen so that it lays within the area of subsequent en bloc resection. Improper biopsy has been shown to be associated with greater morbidity, complications and changes in clinical course and outcome.17 To prevent tumor seeding, biopsy can be obtained by using a coaxial needle with its tip at the outer margin of the tumor through which the core biopsy needle can be placed within the tumor. An adequacy rate of 93% and an accuracy rate of 95% have been reported with FNA and core needle biopsies.16
In case of a malignant retroperitoneal soft tissue tumor, needle biopsy should not be performed routinely because of the potential danger of transperitoneal tumor spread and track seeding. Exceptions include suspected intra-abdominal nodal involvement in lymphoma and germ cell tumors and for soft tissue tumors where preoperative chemotherapy or radiation is to be used.15
The histologic grade of soft tissue sarcoma best predicts its biologic behavior and is determined by four factors: mitotic index, degree of cellularity, intratumoral necrosis and degree of nuclear anaplasia. A soft tissue sarcoma is graded as low-grade or high-grade malignancy on histopathology. High-grade soft tissue sarcomas can be moderately differentiated, poorly differentiated, or undifferentiated.
Key Points Histopathologic considerations
• According to the WHO, more than 50 histologic subtypes of soft tissue sarcomas exist.
• Needle aspiration or core biopsy under US, CT, or MRI guidance or incisional or excisional biopsy of a soft tissue sarcoma can be performed for histologic diagnosis.
• Histopathologically, a soft tissue sarcoma may be low-grade or high-grade. A high-grade tumor may be moderately differentiated, poorly differentiated, or undifferentiated.
Clinical Evaluation
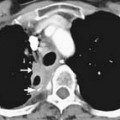
Soft tissue sarcomas usually present as gradually enlarging, painless masses. Diagnosis of soft tissue sarcoma relies on clinical examination, imaging and histologic analysis. Clinical examination and imaging define tumor relationship with adjoining structures.18–20 Because they are more superficial, soft tissue tumors in distal limbs and head and neck tend to be small; those in thighs, buttocks, and retroperitoneum can be huge.
When evaluating a patient with a soft tissue mass, one must ascertain how long the mass has been present; whether the mass is slow- or fast-growing; the presence of local pain and tenderness; constitutional symptoms; history of prior surgery, radiation, or trauma to the area; and recent use of anticoagulants. The growth rate of a soft tissue sarcoma can often vary with aggressiveness of tumor. However, slow growth does not always imply benignity. It is not uncommon for epithelioid sarcoma to be mistaken for a benign tumor because of its small size, thereby leading to improper management. The presence of local pain, seen in about one third of patients with rapidly growing high-grade tumors, usually indicates poor prognosis.18 Discoloration of overlying skin or variation in size of the soft tissue mass with activity or palpation favors hemangioma. Regional lymph nodes should always be examined for metastatic spread, even though lymphangietic spread of a soft tissue sarcoma is uncommon.
Most soft tissue sarcomas usually expand centrifugally and tend to have a peripheral pseudocapsule of compressed normal soft tissue. However, this pseudocapsule is often infiltrated by tumor.21 As a soft tissue sarcoma grows, it follows the path of least resistance and tends to remain confined to the compartment of origin. Such an intracompartmental soft tissue tumor is bounded by natural anatomic barriers, such as fascial septa, tendon, ligament, cortical bone, articular cartilage and joint capsule. Larger tumors can cause symptoms secondary to increased pressure/stretch on adjoining neurovascular structures. This can result not only in pain but also in paresthesias and edema. With highly aggressive soft tissue tumors, satellite tumor foci (skip metastases) may be found frequently beyond the peritumoral reactive zone within the compartment of origin.21
The presence of a pseudocapsule at the periphery of a soft tissue sarcoma does not confer benignity. A well-marginated soft tissue tumor with a pseudocapsule can be malignant, whereas an infiltrating soft tissue tumor with ill-defined borders, such as desmoid tumor, rarely spreads. Size of a soft tissue tumor may not always distinguish a benign from a malignant soft tissue tumor, as has been pointed out earlier. However, in general, a soft tissue mass smaller than 3 cm in diameter tends to be benign with a positive predictive value of 88%, whereas a soft tissue mass larger than 5 cm in diameter indicates malignancy with sensitivity of 74%, specificity of 59% and accuracy of 66%.22 Extracompartmental extent, involvement of adjacent bone, and encasement of neurovascular bundle are insensitive signs of malignancy because these can also be seen with benign soft tissue tumors, such as hemangioma, desmoid and pigmented villonodular synovitis. A benign soft tissue lesion, such as fibromatosis, may be aggressive in behavior, whereas a well-differentiated liposarcoma is slow-growing. In some cases, both benign and malignant soft tissue tumors may be seen in the same patient. For instance, in a patient with neurofibromatosis, numerous benign soft tissue neurofibromas may coexist with a neurofibrosarcoma, and sometimes, it may be difficult to distinguish between the two.
Key Points Clinical evaluation
• Soft tissue sarcomas present as a localized palpable mass with variable pain and tenderness.
• A soft tissue sarcoma is usually hard in consistency, and when large, it may produce symptoms by compression of adjoining neurovascular bundle.
• When the sarcoma is near a joint, the patient may have limitation in range of motion.
• The size of a tumor does not determine whether it is benign or malignant.
• A rapidly growing tumor usually indicates sarcoma.
• A painful, rapidly growing soft tissue tumor tends to have a poor prognosis.
• Pseudocapsule around a soft tissue sarcoma often harbors malignancy.
Classification and Staging
Soft tissue is derived from mesenchyme and consists of skeletal muscle, fat, fibrous tissue, blood vessels and neurovascular tissue. Soft tissue sarcomas (Greek, sarx means “flesh”) are designated on the basis of the adult tissue they resemble microscopically, and not necessarily on the tissue in which they originate.2 For instance, lipoma designation does not mean that the tumor arose from adipose tissue; instead, it means the tumor contains tissue resembling mature fat. Dedifferentiated soft tissue sarcoma contains poorly differentiated mesenchyme on microscopy and, thus, lacks a specific designation to the tumor. More sophisticated immunohistochemical stains and genetic markers are now available to further classify these undifferentiated soft tissue sarcomas. Only a few of the genetic aberrations that occur in soft tissue sarcomas are congenital; most are acquired spontaneously.23
Staging refers to the evaluation of local and distant spread of tumor and has multiple purposes. It provides a standardized method for determining extent of disease, allows assessment of prognosis, and serves to guide initial treatment decisions. The revised seventh edition of the American Joint Commission on Cancer (AJCC) staging system that took effect on January 1, 2010, does not directly take into consideration the histologic subtype, known to be especially important for predicting tumor behavior and metastatic risk. Rather, parameters such as tumor cellularity, differentiation, pleomorphism, mitotic rate, and necrosis are used to assign tumors into one of three possible tumor grades (low-, intermediate-, and high-grade) that indirectly predict a cancers phenotype. (AJCC official web site).
This GTNM system (based on grading, tumor, node and metastases) is shown in Table 37-2.24 However, these criteria do not apply to all soft tissue sarcomas, such as visceral sarcomas and Kaposi’s sarcoma. The recent staging classification has undergone several critical changes. For example, several soft tissue sarcoma subtypes such as gastrointestinal stromal tumors (GISTs), desmoid fibromatosis, and uterine sarcoma, previously included within the general soft tissue sarcoma staging system, now have their own respective staging classification. Conversely, dermatofibrosarcoma protuberans, angiosarcoma, and extraskeletal Ewing’s sarcoma are newly included with the soft tissue sarcoma staging system.
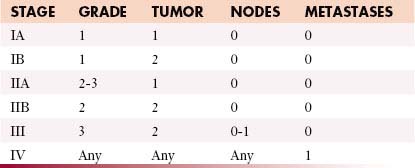
Another change is that tumor depth, previously described as superficial or deep, is no longer used within the latest seventh edition, and the three-tiered grading method advocated by the FNCLCC (Fédération Nationale des Centres de Lutte Contre le Cancer)25 is now preferred over the four-tiered GTNM system (Table 37-3).
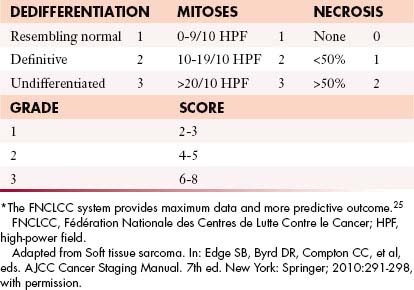
Tumor Characteristics: GTNM System
The most important determinants of staging of extremity soft tissue sarcoma are histologic grade and size; both have similar prognostic value26,27 (see Table 37-3). The N stage refers to regional node involvement. Metastatic spread with soft tissue sarcomas is generally hematogenous; however, in fewer than 5% of cases, metastatic spread may occur via lymphatics. The most common tumors that metastasize to regional lymph nodes are synovial sarcoma, epitheloid sarcoma, clear cell sarcoma, rhabdomyosarcoma, and angiosarcoma.18 The M stage refers to local or distant metastases. Metastatic potentials for low-grade, intermediate-grade, and high-grade soft tissue sarcomas are 5% to 10%, 25% to 30%, and 50% to 60%, respectively.
In general, staging of soft tissue sarcomas is as follows:
Although the staging system continues to evolve, significant challenges remain to be solved.27 Soft tissue sarcomas of extremities, head and neck, viscera, and retroperitoneum are all staged together, regardless of different surgical approach and outcomes.5 In addition, small size alone is not always determinant of actual biologic behavior of soft tissue tumors. For instance, a small epitheloid or synovial sarcoma often disseminates, whereas a much larger well-differentiated liposarcoma rarely does.
Spectrum of Soft Tissue Sarcomas
The WHO classification of soft tissue sarcomas provides uniformity of tissue diagnosis3 (see Table 37-1). However, this classification is incomplete because it does not include neurogenic sarcomas. In addition, at present, atypical lipoma and well-differentiated liposarcoma, having no potential for distant metastases, are considered histopathologically the same tumor, and not different neoplasms, whereas MFH is now designated as undifferentiated high-grade pleomorphic sarcoma.28,29
A modified WHO classification of soft tissue sarcomas is given in Table 37-1. The most common soft tissue sarcomas in adults are high-grade pleomorphic sarcoma (formerly MFH) 28%, liposarcoma 15%, leiomyosarcoma 12%, synovial sarcoma 10%, and malignant peripheral nerve sheath tumors 6%, whereas rhabdomyosarcoma is the most common soft tissue sarcoma in children.18


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


