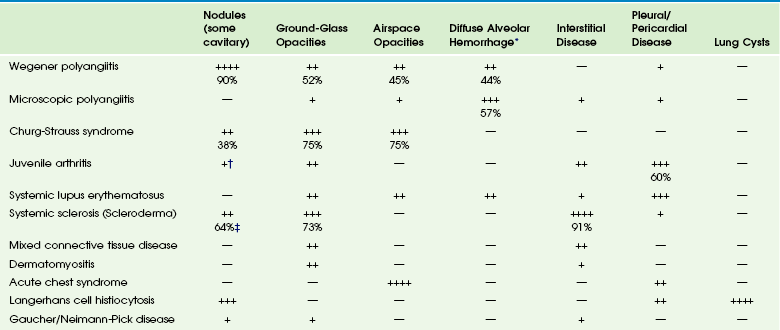
Chapter 57 Pulmonary involvement is a frequent component of systemic illness. Pediatric patients often present for chest imaging before the diagnosis of a specific systemic disorder has been made. Therefore, the radiologist plays an important role in directing the diagnostic workup by recognizing that the pulmonary findings reflect an underlying condition. The first imaging study in pediatric patients with respiratory symptoms is usually chest radiography. In some patients such as those with pulmonary edema or sickle cell disease, radiography shows the pulmonary findings adequately to continue appropriate clinical care. In many patients, however, chest radiography is nonspecific, provides initial clues to the diagnosis, or both, prompting further investigation with computed tomography (CT) of the chest. In recent years, the advent of multidetector CT and controlled ventilation techniques (see Chapter 49) has dramatically improved the detailed assessment of lungs in children with systemic disorders. Vasculitis and Collagen Vascular Disease Although medium- and large-vessel vasculitides more commonly affect mediastinal vasculature structures and rarely the lung parenchyma, small-vessel vasculitides are the most likely to cause pulmonary parenchymal disease. Pediatric patients with pulmonary vasculitis typically present in their teen years. Of these disorders, Wegener polyangiitis (WP), previously known as granulomatosis, is the most common one in children. Microscopic polyangiitis (MPA) and Churg-Strauss syndrome (CSS) are rarely seen in children. Pulmonary nodules and airspace opacities are typically seen on imaging studies.1–3 Pulmonary involvement, most commonly interstitial changes, may be seen in children with collagen vascular diseases (CVD) such as juvenile arthritis, dermatomyositis, systemic sclerosis (scleroderma), systemic lupus erythematosus (SLE), and mixed connective tissue disease. Pulmonary involvement is less common in children than in adults. Pulmonary disease is seen more frequently in children with systemic sclerosis (59% to 91%) than in the other CVDs, and it is associated with significant morbidity and mortality.4,5 A recent study found that abnormal pulmonary function tests (PFTs) were correlated with the severity of abnormalities on high-resolution CT (HRCT). Thus PFTs provide a monitoring tool to identify children who would benefit from further lung disease evaluation with HRCT.5 Symptomatic lung disease is significantly less common in juvenile rheumatoid arthritis and SLE, reported in only 5% of patients.6 Both the vasculitides and CVDs may cause pulmonary renal syndrome, which is the association of both pulmonary hemorrhage and glomerulonephritis. It is often seen in WP and SLE.1,2 Pulmonary hemorrhage may be seen in both the vasculitides and in CVDs (e-Fig. 57-1) and has a high morbidity and mortality (50% to 90%).1,7 e-Figure 57-1 Pulmonary hemorrhage in a teenage boy with pulmonary vasculitis resulting from collagen vascular disease. Etiology: WP is, by far, the most common of the pulmonary vasculitides seen in children and typically presents with a triad of necrotizing granulomatous lesions in both the upper and lower respiratory tract, as well as glomerulonephritis. MPA is a nongranulomatous necrotizing vasculitis almost always seen with glomerulonephritis. CSS (also known as allergic granulomatosis and angiitis) typically presents with asthma and blood eosinophilia but is rare in children.1 Most of the pulmonary vasculitides are immunologically mediated. WP, MPA, and CSS are associated with antineutrophil cytoplasmic antibodies (ANCA) and are sometimes referred to as ANCA-associated systemic vasculitides. Imaging: The common imaging findings of pulmonary vasculitis and CVDs are summarized in Table 57-1. The frequent imaging findings of WP are variable sized nodules, followed by ground-glass opacities and air space consolidation; 17% of the nodules show cavitation.8 The nodules are frequently surrounded by a halo of ground-glass opacity, which represents hemorrhage (e-Fig. 57-2 and Fig. 57-3). Airway wall thickening may also be seen, but airway strictures are significantly less common in children (3%) compared with adults (up to 59%).8 Diffuse alveolar hemorrhage (occurring in 44% of pediatric WP) is characterized by lobular or lobar regions of ground-glass opacity or airspace consolidation on CT.9–11 It may also show the “crazy paving” pattern on CT, especially as it evolves.12 Table 57-1 Pulmonary Imaging Findings in Systemic Diseases *Specific imaging findings are a spectrum from ground-glass opacities to airspace consolidation. †Nodules in juvenile arthritis seen in lipoid pneumonia. ‡Seen as subpleural micronodules and were not considered a dominant finding. Data from references 1, 4, 6, 7, and 11; and Feng RE, Xu WB, Shi JH, et al. Pathological and high resolution CT findings in Churg-Strauss syndrome. Chin Med Sci J. 2011;26(1):1-8. Figure 57-3 Wegener polyangiitis. e-Figure 57-2 Wegener polyangiitis. In the case of CVDs, pleural and pericardial effusions are the most common findings in the chest. Pulmonary findings are significantly less common but follow a similar pattern for most of these disorders and include ground-glass opacity and interstitial septal thickening, which may progress to pulmonary fibrosis. Despite normal chest radiographs or only minimal radiographic abnormalities in children with systemic sclerosis, HRCT demonstrates ground-glass opacities, peripheral areas of pulmonary fibrosis, and subpleural micronodules (Fig. 57-4).4 Some authors have reported occurrence of lipoid pneumonia in children with juvenile idiopathic arthritis, not associated with mineral oil ingestion.12 “Shrinking lung syndrome” has been described in SLE and represents decrease in lung volume that manifests radiographically by an elevated diaphragm.7,12,13 Figure 57-4 An 8-year-old girl with systemic sclerosis (scleroderma) diagnosed at age 5 years. Treatment and Follow-up: Treatment for vasculitides and CVDs is primarily aimed at suppressing the immune response, typically with corticosteroids and chemotherapeutic agents. In patients with vasculitis, CVDs, or both, who experience diffuse alveolar hemorrhage, evolving changes may be seen on CT at follow-up. A more linear and interstitial type of pattern may develop with interlobular septal thickening and the appearance of the “crazy paving” pattern. If episodes of hemorrhage recur, this may also progress to interstitial fibrosis.9 Pulmonary involvement with CVD may progress to interstitial pulmonary fibrosis regardless of treatment, with advanced stages showing honeycombing. Pulmonary artery hypertension may also develop with advanced lung disease, especially in systemic sclerosis. Acute chest syndrome (ACS), a pulmonary illness that occurs in up to 50% of children with sickle cell disease (SCD), is characterized by chest pain, leukocytosis, fever, and a new pulmonary opacity. It is the leading cause of death (25%) and hospitalization in all patients with SCD and usually occurs between 2 and 4 years of age.14 ACS also occurs in patients with other sickle hemoglobinopathies.15 When not fatal, it may lead to chronic lung disease (4%) and pulmonary hypertension.16 Etiology: The etiology of ACS is complex and not always known. In a large multicenter study of 671 episodes, causes were found to be microvascular occlusion and infarction (16%), infection (29%), fat emboli from bone marrow infarcts (9%), and unknown in 46%. Chlamydia, Mycoplasma, and viral agents were the most common pathogens in cases caused by infection. One third of patients presented with symptoms of long bone pain caused by vasoocclusive crisis and developed ACS 2 to 3 days later.17 Imaging: Radiographic findings of ACS are nonspecific but, by definition, include the presence of a pulmonary opacity (e-Fig. 57-5). Opacities were noted in the lower lobes in about 90%, and pleural effusion was noted in 55% of cases in a large multicenter study (see Table 57-1).17 e-Figure 57-5 Sickle cell disease with acute chest syndrome. Although CT is not generally used in the setting of ACS, chronic sickle cell lung disease has been studied by using HRCT. Abnormal findings most pronounced at the lung bases include parenchymal bands, interlobular septal thickening, architectural distortion, and traction bronchiectasis. Honeycombing is unusual, in contrast to other types of pulmonary fibrosis.18 Treatment and Follow-up: Therapy for ACS includes hydration, analgesia, respiratory support (including bronchodilators), broad-spectrum antibiotics, transfusion therapy, and sometimes corticosteroids.19 Imaging follow-up is primarily dictated by clinical progression, improvement, or both and may be accomplished by using radiography. Langerhans cell histiocytosis (LCH) is now the preferred term describing the condition showing a proliferation of a group of histiocytes known as Langerhans cells. Langerhans cells are of myeloid dendritic cell origin and contain characteristic Birbeck bodies on histology.20 The peak age at initial diagnosis is 1 to 3 years, and most patients present with osseous lesions.20 LCH is currently classified according to the extent of involvement, whether it involves a single site (better prognosis) or multiple sites (higher risk of long-term problems). The involvement of “risk organs” (liver, spleen, lung, bone marrow) indicates a poorer prognosis and demands more aggressive treatment.21 Pulmonary involvement is reported to be present in 23% to 50% of children with multisystem LCH.12,20 The mean age of patients with lung disease was 11.9 months in one study. Disease-free survival in these patients was 69%, with 3 out of 4 deaths occurring when “risk organs” other than the lung were involved.22 Although lung disease was traditionally considered a poor prognostic indicator in LCH, recent studies have shown that it does not adversely affect outcome. Primary pulmonary LCH (single site) is rare in children and is typically seen in young adult smokers.21,22 Etiology: The etiology of LCH is not known. Most investigators believe it to be an immune-mediated condition, in which the Langerhans cells accumulate and cause an inflammatory reaction. In the lungs, destructive granulomatous lesions occur in the interstitial tissues, bronchial and bronchiolar epithelium, and subpleural septa that may eventually lead to cystic lesions.20,21 Pulmonary fibrosis occurs later in 10% of children and may demonstrate multiple small cysts giving the lung a honeycomb appearance.23 Imaging: Radiologic findings of LCH vary widely with the extent of the disease and are summarized in Table 57-1. Initial chest radiography may be normal. Small nodules and cysts may be seen, with upper lobe involvement equal to or more extensive than lower lobe involvement (Fig. 57-6). The reticular appearance noted on chest radiography is often from multiple small cysts.20,24 LCH is the most common cause of acquired extensive cystic lung disease in children (e-Fig. 57-7). Spontaneous pneumothorax occurs in about 11% of patients with pulmonary involvement and may be the first indicator of pulmonary disease.25 However, it may be also a manifestation of more advanced disease, as in adolescents who more often have large coalescent cysts (see e-Fig. 57-7).12 Figure 57-6 Langerhans cell histiocytosis of the lungs in a 2.5-month-old boy who initially presented with a skin rash. e-Figure 57-7 Progressive advanced changes in Langerhans cell histiocytosis (LCH) of the lungs. CT is indicated for neonates with LCH and any patient with abnormalities on chest radiography.21 HRCT findings in LCH are characterized by small nodules (with or without cavitation) with upper and middle lobe predominance (e-Fig. 57-8).12,20,26 The pleura may be thickened and sometimes replaced by a thick layer of granulomatous tissue. However, pleural effusions are rare.27 Mediastinal and hilar adenopathy is also rare in children with pulmonary LCH.20,24 e-Figure 57-8 A patient with known diagnosis of Langerhans cell histiocytosis with relapse of disease with osseous lesion in the scapula (not shown). Treatment and Follow-up: Treatment of pulmonary LCH depends on the extent of disease and whether “risk” organs are involved. Generally, the first line of treatment is corticosteroids, vinblastine, or both, with duration of therapy being determined by response. Radiation therapy has fallen out of favor except for treating unstable bone lesions.21 Imaging follow-up for pulmonary LCH depends on clinical symptoms and is best performed with CT. Etiology: Gaucher disease is caused by a deficiency of the enzyme glucocerebrosidase that results in an accumulation of glucocerebroside in phagocytic cells. Pulmonary involvement is most common in patients with neuronopathic types. Neimann-Pick disease is caused by a deficiency of sphingomyelinase, resulting in accumulation of sphingomyelin. Progressive pulmonary fibrosis has rarely been reported in Neimann-Pick disease.28 Imaging: A diffuse reticular lung pattern may be seen on chest radiography with Gaucher disease.29 Niemann-Pick disease may produce similar findings.27 Lipoid pneumonia has been described in patients with Neimann-Pick disease.30 Imaging findings are summarized in Table 57-1. Treatment and Follow-up: Visceral involvement may improve with enzyme replacement therapy in Gaucher disease, and this therapy may offer an alternative to lung transplantation. The current treatment for Neimann-Pick disease is whole lung lavage.28,30 CT is more sensitive than plain radiography for imaging follow-up, when clinically warranted. In the pediatric population, cardiogenic edema usually presents in infants less than 6 months of age and is the result of congenital heart disease (CHD) (Figs. 57-9 and 57-10). In an older child, cardiomyopathy may be the cause of cardiogenic edema. Affected infants often present with nonspecific symptoms, including feeding difficulties, grunting, diaphoresis, wheezing, and retractions.31 Rarely, pulmonary edema may be the presenting manifestation of CHD. Figure 57-9 Pulmonary edema caused by obstructed pulmonary venous return. This 6-week-old infant presented to the emergency room with tachypnea and feeding difficulties. Figure 57-10 Cardiogenic edema. The frequent causes of noncardiogenic pulmonary edema in children include acute respiratory distress syndrome (ARDS), near-drowning, neurogenic pulmonary edema, renal disease, and upper airway obstruction. Other less common causes of noncardiogenic pulmonary edema include aspiration pneumonia, hydrocarbon pneumonitis, smoke inhalation, and drug reactions.32 Etiology: Cardiogenic pulmonary edema occurs significantly less frequently in children than in adults. The common underlying causes for cardiogenic pulmonary edema include left-to-right shunting lesions, left ventricular outlet obstruction, obstruction of pulmonary venous return, or cardiomyopathy (e-Fig. 57-11).31 Cardiogenic pulmonary edema progresses in severity as pulmonary capillary wedge pressure increases. e-Figure 57-11 Dilated cardiomyopathy. Imaging: On chest radiography, cardiomegaly is usually found except in instances of pulmonary venous obstruction, in which interstitial edema is present in the absence of cardiac enlargement (see Fig. 57-9). Early findings in interstitial edema in children with cardiogenic pulmonary edema are indistinct vascular margins and bronchial wall thickening. Interlobular septal thickening (septal lines or Kerley B lines) and interlobar fissural thickening are also often seen. In infants and young children, fissural thickening is often more easily recognized than Kerley lines and is therefore an important clue for diagnosing cardiogenic pulmonary edema in children (see Fig. 57-9). An indirect radiographic finding often seen in children with CHD is hyperinflation. This may be a pitfall for the imager if he or she attributes the findings of peribronchial thickening and hyperinflation to airways disease and does not recognize them as subtle findings of interstitial edema (see Fig. 57-9). Alveolar edema usually occurs after development of interstitial edema and is the most recognizable radiographic finding of pulmonary edema (see Fig. 57-10 and e-Fig. 57-11). The classic appearance of acute alveolar edema is a central or “butterfly” distribution of airspace disease, with central edema and sparing of the lung periphery. Pleural effusions are also usually present (see e-Fig. 57-11). Although not commonly used to diagnose pulmonary edema, CT is sensitive for the detection of pulmonary edema of any etiology. CT shows peribronchial cuffing, septal lines, ground-glass opacities, and air space consolidation in order of increasing severity (e-Fig. 57-12).33,34 e-Figure 57-12 Glomerulonephritis. Treatment and Follow-up: Treatment for cardiogenic pulmonary edema is generally supportive and includes corrective or palliative treatment for CHD. Diuretics and inotropes may be used for cardiac dysfunction. Ventilator support is provided, if needed. Extracorporeal membrane oxygenation and other types of ventricular assist devices are now being used more frequently in the setting of pediatric cardiac failure.35 Typically, imaging follow-up with chest radiography is adequate, although CT may be used to clarify the vascular anatomy. Etiology: Many episodes of noncardiogenic pulmonary edema progress to ARDS. ARDS accounts for 1% to 3% of pediatric intensive care admissions, and the mortality in various series ranges from 40% to 60%.32 Sepsis, near-drowning, pneumonia, and smoke inhalation are the most frequent antecedents to pediatric ARDS. In most cases, ARDS progresses through stages of immediate lung injury, exudative alveolitis, fibroproliferative repair, and, in survivors, recovery. Ventilation–perfusion imbalance leads to varying degrees of hypoxemia.32
Systemic Conditions with Lung Involvement
Specific Systemic Diseases
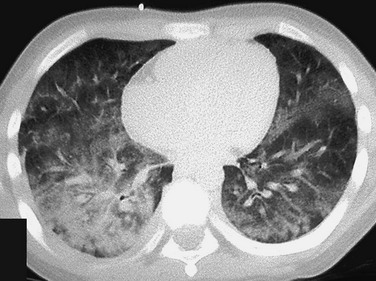
The patient presented with hemoptysis. A computed tomography scan shows diffuse but somewhat patchy ground-glass opacification in both lungs.
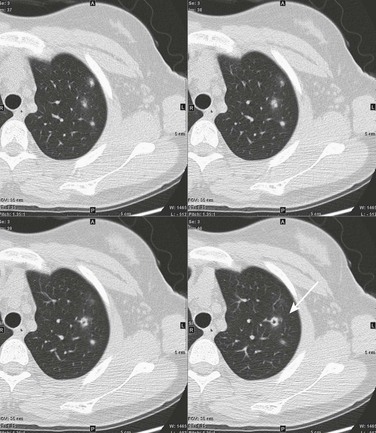
This 16-year-old girl had a history of vasculitic skin lesions and headache. Computed tomography (CT) of the sinuses revealed inflammatory changes (not shown). Axial CT images of the chest show left upper lobe cavitary lesion (arrow) with surrounding ground-glass halo as well as several solid pulmonary nodules, also with surrounding halos of ground-glass opacity. Lung biopsy was positive for Wegener polyangiitis.
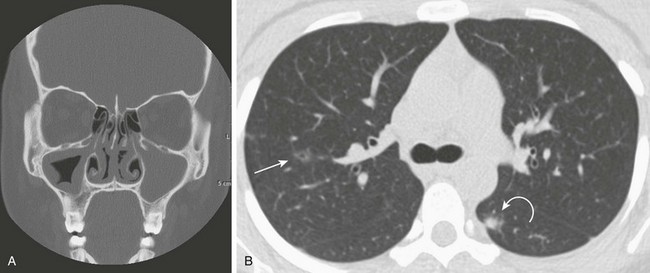
This 16-year-old girl with a history of chronic sinusitis presented with hematuria, acute renal failure, and sinusitis. She was eventually found to have Wegener polyangiitis. A, A coronal computed tomography (CT) image shows mucosal thickening in the right maxillary sinus and opacified left maxillary sinus. B, An axial CT image in lung windows through the upper lobes show a small thin-walled cavitary lesion in the right upper lobe (arrow). Also shown is a focal nodule with a halo of ground-glass opacity in the superior segment of the left lower lobe (curved arrow).
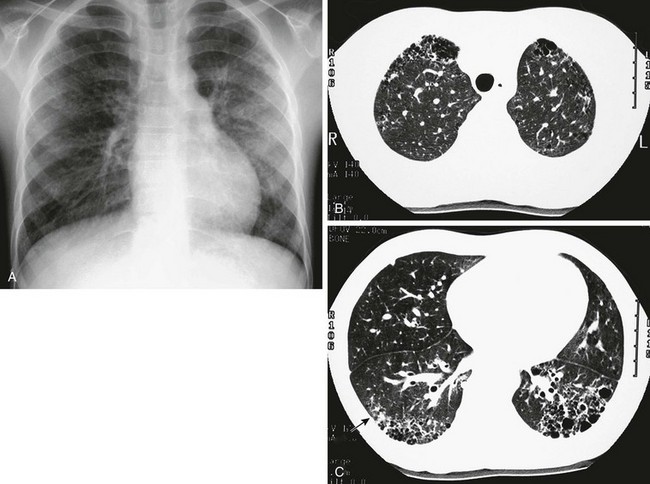
A, The frontal chest radiograph shows mild increase in perihilar linear opacities. B, High-resolution computed tomography (HRCT) of upper lung zones shows thin-walled, small cystic areas anteriorly in both lungs and a few small scattered subpleural lucencies and opacities more posteriorly. C, HRCT through lower lung zones shows honeycomb pattern dependently in both lungs. More lateral area in right lung shows more ground-glass opacity (arrow) that may represent active disease.
Sickle Cell Disease
An 8-year-old boy with sickle cell disease who presents with fever. A and B, Posteroanterior and lateral chest radiographs show dense airspace consolidation in the left lower lobe with small pleural effusion.
Langerhans Cell Histiocytosis
A, An anteroposterior chest radiograph shows a coarse reticulonodular interstitial pattern. B, An axial computed tomography image in a lung window shows the coarse reticulonodular interstitial pattern with some small thin-walled cysts seen in the anterior lung bilaterally.
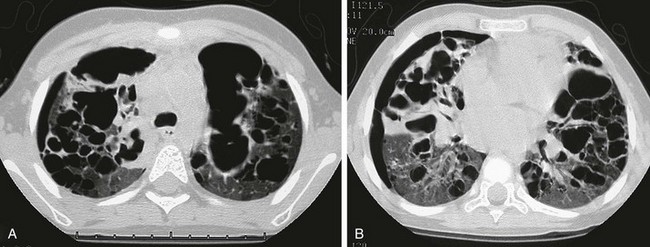
This child first presented at age 2 years with LCH involving both temporal bones and cystic lung disease. The patient was treated with chemotherapy and returned at age 4 years with increasing respiratory distress. The chest radiograph (not shown) showed a marked interval increase in bilateral cystic lung changes and suggested a possible small right pneumothorax. A and B, High-resolution computed tomography shows that multiple cysts of varying sizes have largely replaced much of the lung parenchyma; loculated right pneumothorax was confirmed. Within 3 weeks, the patient developed a large left pneumothorax and died of this cystic lung process 1 week later.
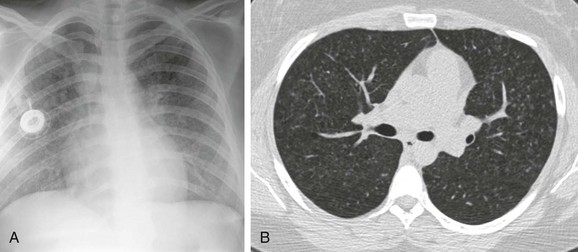
A, The chest radiograph shows nodular interstitial abnormality representing pulmonary histiocytosis. B, An axial chest computed tomography (CT) image shows more clearly the small interstitial nodules and tiny thin-walled cysts in the upper lobes, characteristic CT findings of histiocytosis.
Gaucher Disease and Niemann-Pick Disease
Pulmonary Conditions Occurring with Generalized Systemic Illness
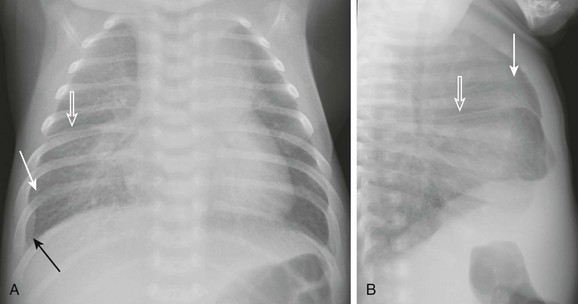
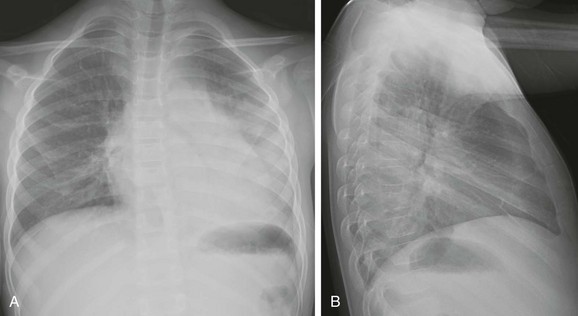
A and B, Anteroposterior and lateral chest radiographs show normal heart size and subtle interstitial edema characterized by hyperinflation, septal lines (white arrows), fissural thickening (open arrows), and tiny right pleural effusion (black arrow). Diagnosis of cor triatriatum was made with echocardiography.
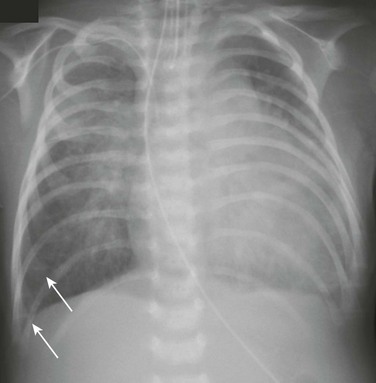
A 5-week-old male infant with known ventricular septal defect presenting with tachypnea and respiratory distress requiring intensive care unit admission. An anteroposterior chest radiograph shows cardiomegaly, hyperinflation, and pulmonary edema characterized by perihilar airspace opacity, indistinct hilar vessels, and septal lines (arrows).
Cardiogenic Pulmonary Edema
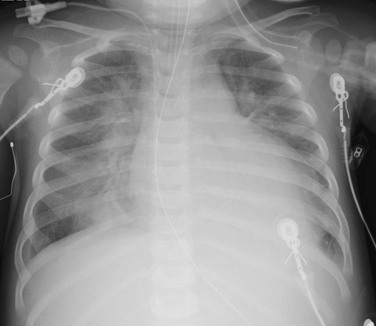
A 14-month-old female presented with 24 hours of increased work of breathing and wheezing. The portable chest radiograph shows characteristic cardiomegaly, perihilar and basilar distribution of airspace opacity caused by pulmonary edema, and a small right pleural effusion.
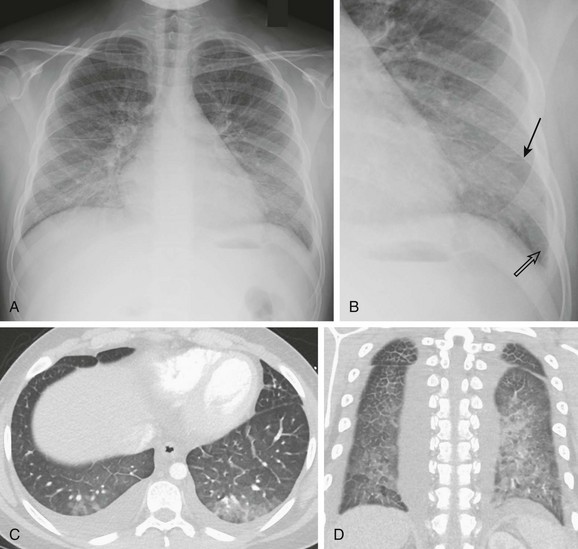
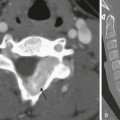
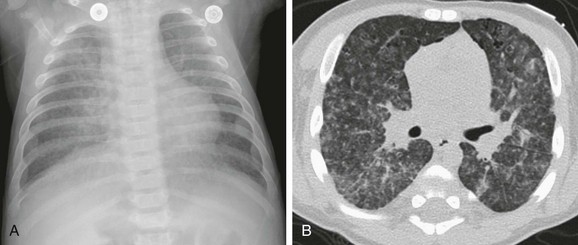
A 13-year-old male with history of asthma presented with 5 days of shortness of breath and was found to be hypoxic. A and B, Posteroanterior chest radiograph and magnified view show pulmonary edema with interstitial septal lines (arrow) and tiny left pleural effusion (open arrow). C and D, Axial and coronal computed tomography images depict typical findings of pulmonary edema showing interlobular septal thickening, dependent ground glass opacity, and bilateral small pleural effusions. The child was diagnosed with postinfectious glomerulonephritis.
Noncardiogenic Pulmonary Edema
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree