Fig. 1
The spectral layout of four different IFC systems we have successfully used to implement versions of our method. In all cases, the excitation lasers and approximate emission channel widths are shown with major fluorochrome examples to aid panel design. Although the IS100 offers limited flexibility, we have successfully tracked division with CTV, identified mitotic cells with PI/PH3 and measured one fate-determining signal within these cells [17]. The newer generation systems offer greater experimental flexibility and data content
ImageStream 100 (IS100): 10-bit CCD camera, dynamic range 0–1024, 6 camera channels, and up to 5 of these can be fluorescent stains plus side scatter, fixed at 40× magnification.
FlowSight (FS), 12-bit CCD camera, dynamic range 0–4096, 12 camera channels up to 10 of which can be used for fluorescent stains, fixed at 20× magnification.
ImageStreamX MKI or MKII (ISXMKI/II): 12-bit CCD camera, dynamic range 0–4096, 6 or 12 camera channels of which 5/10 of which can be used for fluorescent stains depending on configuration, 20, 40, or 60× magnification with multi-mag upgrade otherwise fixed at 40×.
All systems should possess 488 nm, 405 nm, and 654/642 nm excitation lasers, with a 561 nm laser optional (not available for the IS100).
All systems should be fully ASISST calibrated with additionally quality control performed using a multi-level fluorescent microsphere set. Particular attention should be paid to the resolution of peaks 2 and 3 (dim 1 and dim 2) at laser powers used to excite experimental samples.
2.2.
A conventional flow cytometer (CFC) system with an analogous excitation laser and detector configuration to the IFC systems listed above (see Note 1 ).
2.3.
Media and buffers:
2.3.1.
Primary T cell/Jurkat culture media: RPMI supplemented with 10 % fetal bovine serum (FBS), 100 units of penicillin/streptomycin, 2 mM glutamine, and 50 μM 2-mercaptoethanol (2-Me).
2.3.2.
B cell culture media: RPMI supplemented with 10 % FBS, 50 μM 2-Me, 25 mM HEPES, 2 mM GlutaMAX, and 10 U/ml penicillin/streptomycin.
2.3.3.
Wash/stain buffer: PBS supplemented with 2 % FBS.
2.3.4.
Permeabilization buffer: Wash/stain buffer supplemented with 0.1 % Triton-X 100.
2.4.
CellTrace Violet™ (Cat No A10198: Life Technologies): There are a number of commercially available fluorescent dyes that are able to record the division history of single cells through dye dilution [18]. In general terms there are two categories of reagents, succinimidyl esters and lipophilic dyes (see Notes 2 – 4 ). CTV is a succinimidyl ester.
2.4.1.
CTV is supplied as a lyophilized powder (stored at −20 °C till the manufacturer’s expiration date) that is reconstituted in 20 μl of DMSO to a stock concentration of 5 mM. Working concentrations range between 1 and 10 μM and should be determined empirically for each cell type. Aliquots of reconstituted dye can be stored at −20 °C and freeze-thawed once. It is excited by the 405 nm laser and emits at 450 nm (Fig. 1, lower panel).
2.4.2.
The exact activation reagents required depend on the immune cell type being studied for ACD. All culture, activation and staining/washing steps are done in 96 well round bottom plate or 24-well flat-bottomed plates (various suppliers).
2.4.3.
F5 transgenic T cell activation reagents:
NP68 peptide (synthesized in house): The stimulatory peptide for the F5 receptor.
Anti-CD3 antibody (clone 145-2C11): A generic T cell activator.
Anti-CD28 antibody: A co-stimulatory signal.
Recombinant ICAM-1: Thought to be essential for driving ACD.
2.4.4.
B cell activation reagents:
CD40 ligand. Mimics effect of a helper T cell signal.
Anti-IgM antibody: Stimulated through the B cell receptor.
Interleukin-4: T cell-derived cytokine.
2.5.
Antibodies against intracellular and extracellular targets should be chosen depending on the molecules/cell cycle phases under investigation. For T cell studies CD69 or Ki67 are used to identify cells that have been triggered to divide. This allows one to determine the best harvest window in terms of the greatest number of cycling cells [12]. Antibodies should be directly conjugated when possible with fluorochromes compatible with the overall panel and spectral properties of the IFC instrument. Species, clones, and cross-reactivity should also be considered (see Note 5 ). For particular markers/antigens we strongly recommend using the indicated fluorochromes (shown in bold type). For markers where no particular fluorochrome is recommended, one can be selected that fits into a free channel on the IFC system used (see Fig. 1). Examples of some marker/fluorochrome combinations we have used are:
2.5.1.
Anti-Ki67: Used for measuring cell cycle entry.
2.5.2.
Anti-CD69 (clone H1.2F3): Denotes commitment to proliferation.
2.5.3.
Anti-MPM2 (clone MPM2), for example FITC or Cy5 conjugated (see Note 5 ): Identifies all stages of mitotic cells.
2.5.4.
Anti-PKC zeta goat anti rabbit (cat no SC-216-G, Santa Cruz, USA): Strongly implicated as an asymmetrically inherited fate marker.
2.5.5.
Donkey anti-goat PE (CH3) or AlexaFluor-647, (CH11): To detect the unconjugated PKC zeta goat antibody (see Note 5 ).
2.5.6.
Anti-CD8 (clone 53-6.7): A possible marker for T cell ACD.
2.5.7.
Anti-CD25 (clone PC61.5): A possible marker for T and B cell ACD.
2.5.8.
Anti-CD86 (clone GL1): A possible marker for B cell ACD.
2.5.9.
Anti-IA/IE (clone M5/114.15.2): A possible marker for B cell ACD.
2.6.
FC-block reagent (anti CD16/32 clone 2.4G2): Used to block nonspecific binding of antibodies via their Fc regions.
2.7.
Near-infrared live/dead (NIRLD) fixable dye (CH12): Note that this dye is not compatible with an IS100, but can be used on a single-camera ISx system in CH6. BF illumination can also feature in this channel as NIRLD signal from dead cells will cause “white out” in the BF image and dead cells can be gated out using the gradient RMS feature for focus along with defocused cells.
2.8.
2.9.
Propidium iodide/7-AAD: Both are available from several commercial sources. Stocks are made up at 50 μg/ml in DI-H2O.
2.10.
Siliconized 1.5 ml microfuge tubes or protein “low-bind” version.
2.11.
Copies of IDEAS (Amnis), FlowJo (Treestar Inc.) or equivalent FCS-based software analysis package: Image J (open-source image analysis package, NIH, USA). This protocol assumes a good working knowledge of these software packages. For IDEAS, users should understand compensation, masking, and feature creation. Users should also know how to export feature values and raw images for analysis in third party software. More information can be found in the respective user guides.
3 Methods
3.1 CTV Labeling
3.1.1 Labeling Primary T and B Cells with CTV
Cells are washed once in complete culture media and resuspended for cell counting. Centrifugation conditions are 1200 rpm/300 × g for 3–5 min followed by the removal of all supernatant (hereby referred to in this protocol as “washing cells”). Cell viability should also be determined using a dye exclusion method such as trypan blue.
CTV staining solution is made up in protein-free sterile PBS and pre-warmed at 37 °C prior to use. Concentrations for primary T and B cells should be determined empirically based on viability and uniformity of the CTV signal [19]. Labelling solutions should be made up at 2× concentrations. Typical final concentration used for T and B cell labeling is 2.5 μM.
Cells should be spun down and the supernatant carefully aspirated away as any residual protein can compete out the intracellular CTV labeling. The cell pellet should be resuspended in one volume of protein-free PBS so that once the 2× labeling solution is added at an equal volume, the final cell concentration should range between 1 and 3 × 106/ml. The cells should then be incubated at 37 °C for 10 for 5 min (B cells) or 10 min (T cells).
After incubation, neat FBS should be added to the labeling solution to a final concentration of 10 % and incubated for a minimum of 5 min at room temperature. This will terminate intracellular labeling by binding any remaining free dye from the solution.
Cells should then be washed once in full media and resuspended for counting and viability assessment. An aliquot of cells should also be analyzed by CFC in order to determine the intensity and uniformity of CTV labeling as this affects division peak resolution (see Notes 2 and 7 ). Cells can then be adjusted to the appropriate density for subsequent culture and activation.
3.1.2 Labeling Cell Lines
Cell lines are labeled with CTV as described above with no further modifications.
In our hands, CTV successfully labels a number of cell lines (e.g., Jurkat/A549); however the labeled width always exceeds the limits for division peak resolution due to cell-intrinsic heterogeneity (see Note 7 ). This requires cells to be sorted prior to culture in order to narrow the input width sufficiently to resolve division peaks. This method is beyond the scope of this protocol and is described in full detail elsewhere [19].
3.2 Culture and Activation Conditions
3.2.1 T Cells
An example of the in vitro T cell activation protocol from our lab has been described elsewhere [12]. However, briefly CTV-labeled T cells are seeded in 24-well plates at a density of 1 × 106/ml in the presence of various activators, including peptide loaded dendritic cells or anti-CD3 (145-2C11, plate bound at 10 μg/ml or soluble at 0.5 μg/ml) with or without plate bound CD28 and/or recombinant ICAM-1 (both at 10 μg/ml). The cells are harvested at the point of cell cycle commitment based on Ki-67/CD69 expression in the CTV-undivided population (see Note 8 ). Typically this tends to be between 32 and 36 h. for MHCI-restricted F5 CD8 T cells [12].
3.2.2 B Cells
CTV-labeled B cells are seeded in 24 or 96 well-flat bottom plates at a density of 1 × 106 cells/ml. To stimulate B cell proliferation we add CD40L at concentrations ranging from 1 to 0.001 μg/ml or anti-IgM at concentrations ranging from 10 to 0.001 μg/ml. In all cases, B cell culture media must be supplemented with 5 ng/ml IL-4. In our system, B cell proliferation begins 48 h after stimulation with an optimal harvest time of 72 h.
3.2.3 Culturing Cell Lines for Chemotherapeutic Drug Assays
CTV-labeled cell lines, pre-sorted to optimal input width for division peak resolution, are placed back into culture at a density of 0.5 × 106 cells/ml in complete media with a total minimum cell number of 1 × 106 cells (2 ml) per well per analysis time point.
An aliquot of post-sorted cells (1 × 106 cells) are fixed and stained to determine the input cell cycle distribution. Cells are allowed to recover post-sorting for a minimum of 24 h before addition of any cell cycle modulating compounds.
At the point of drug addition, a time zero sample is taken (1 × 106 cells) and fixed under appropriate conditions as described later in the protocol. As a control, cells are also incubated in the absence of drug for the full culture period. Cells can then be cultured for defined periods in the presence or absence of compound.
To model the effects of drug metabolism and removal, one can also investigate the effects of washing the drug away and re-culturing the cells to determine long-term irreversible effects on proliferation to model the effects of drug clearance. The ability to track cells over multiple cell divisions using CTV (~6 rounds) makes such long-term tracking a viable option compared to other temporal methods. As a control at this stage, a sample of cells is taken and fixed at the point of drug removal. A further control is set up where the presence of drug is maintained throughout the culture period.
3.3 Live/Dead Fixable Dyes (NIRLD)
Harvest cells from culture and wash ×1 in wash buffer. Add 2 μl of LD-NIR per ml and incubate at RT for 10 min. Wash cells again then proceed to extracellular staining.
3.4 Extracellular Antibody Labeling
1.
This stage is only required if surface markers are to be investigated. Furthermore, if the surface epitopes are unaffected by fixative treatment, then all antibodies (extra- and intracellular) can be added post-fixation/permeabilization. All antibodies and dyes should be optimally titrated for use on an IFC system paying particular attention to the relative brightness of all fluorochromes excited by the same laser (see Note 9 ).
2.
Make up the cocktail of extracellular antibodies at the pre-optimized concentrations in stain/wash buffer. Add 100 μl per well to stain 1 × 106 cells. If you are using tandem dyes, be sure to check that they remain stable after fixation. Also pay attention to the fact that tandem dyes will be prone to cross laser excitation (see Note 9 ).
3.
For staining, cells should be seeded in a round-bottomed 96-well plate at a density of 1 × 106 cells/ml. Often we stain 2–3 million cells in total per sample. This will require setting up multiple wells per sample to be pooled at the end of staining.
4.
If required, incubate cells with anti-FC block made up in stain/wash buffer for 10 min, then wash. After removing the supernatant, briefly press plate bottom onto a vortex mixer to break up the cell pellets. This can be done after every wash step.
5.
Add 100 μl of antibody cocktail per well and incubate cells in the dark at RT for at least 1 h. Wash cells twice in stain/wash buffer and incubate with secondary labelled antibodies if required.
6.
Single stained controls for each fluorochrome in the panel should be prepared and processed exactly as experimental samples. Allow for the inclusion of single stains against intracellular targets by preparing these wells in advance. Also set up further control wells containing unlabelled cells at exactly the same cell density as experimental samples. These will serve as the PI compensation single cell control when the dye is added to the samples at the end of the staining process.
3.5 Fixation (See Note 6)
3.5.1 70 % EtOH Fixation
If you are only measuring cell cycle distribution without extensive antigenic staining then 70 % EtOH fixation is optimal as it gives the best DNA profiles. It is also compatible with MPM2 staining for mitotic phases [19].
For fixation, ice-cold 70 % EtOH is added to the pellet and then samples are incubated for a minimum of 1 h at RT. EtOH fixed cells can be kept in fixative for 24–48 h without detrimental effects on the CTV peak resolution.
3.5.2 Formaldehyde Fixation
ACD analysis will require staining for antigens in addition to MPM2; therefore EtOH will likely destroy the epitopes necessitating a switch to FA. We have found that a lower concentration of formaldehyde (0.5 %) work best for DNA and MPM2 staining resolution in cell lines (Fig. 2a) and primary murine B cells (Fig. 2b).
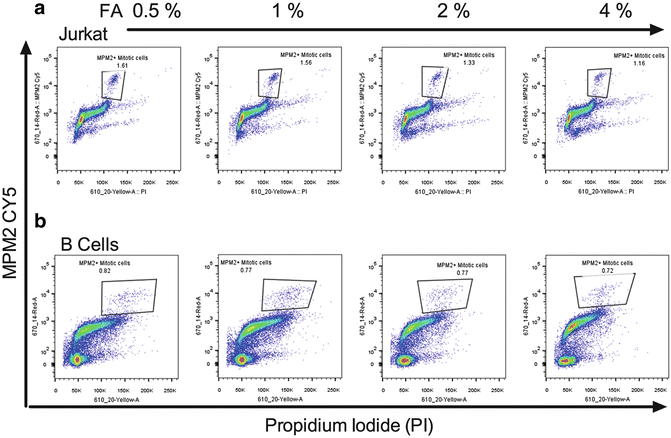
Fig. 2
Identifying mitotic cells by MPM2 staining. (a) Jurkat cells and (b) primary mouse B cells were fixed with the indicated concentrations of FA as outline in Subheading 3 and stained with PI (x-axis) and MPM2 (y-axis)
The cell pellet is first resuspended in one volume of PBS to eliminate clumping. An equal volume of 2× FA solution is then added to a final desired fixative concentration (0.5 %). Incubation should be done at RT for a minimum of 1 h. Cells can be washed out of fixative and stored in stain/wash buffer for up to 24–48 h without loss of CTV peak resolution.
3.6 Intracellular Antibody Labeling
1.
Cells are first permeabilized in 100 μl of permeabilization buffer for a maximum of 5 min, after which 100 μl of wash buffer is added prior to centrifugation. Next add the cocktail of intracellular antibodies made up at appropriate dilutions in wash/stain buffer and incubate at RT in the dark for minimum 1 h. At the same time, stain for any single-stain compensation samples that require antibodies against intracellular antigens.
2.
Wash cells a minimum of two times. If required, incubate with fluorescently labeled secondary antibodies made up in wash/stain buffer for a minimum of 45 min in the dark at RT, after which wash cells once.
3.
The final volume for sample resuspension should not exceed 60 μl (see Note 10 ). Pool multiple wells if needed by adding 60 μl of stain/wash buffer to the first well and transfer to subsequent wells until there is approximately 2–3 × 106 cells in the 60 μl volume. Transfer to a 1.5 ml siliconized, low-bind microfuge tubes.
3.7 DNA Dye Staining
1.
Get Clinical Tree app for offline access

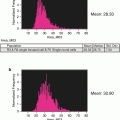
PI concentration should be pre-titrated to avoid camera saturation at a given laser power (see Note 9 ) while still giving well-defined DNA profile based on G1 CVs (Fig. 3a).
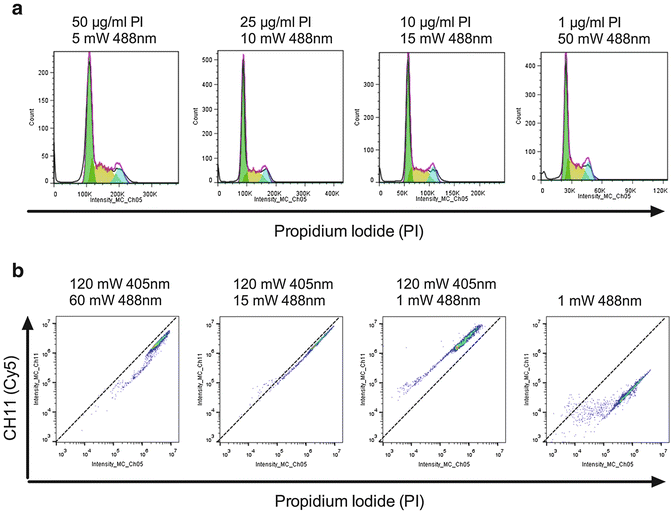
Fig. 3
 Quantitative Functional Morphology by Imaging Flow Cytometry
Principles of Amnis Imaging Flow Cytometry
Quantitative Functional Morphology by Imaging Flow Cytometry
Principles of Amnis Imaging Flow Cytometry
 Detection and Characterization of Rare Circulating Endothelial Cells by Imaging Flow Cytometry
Detection and Characterization of Rare Circulating Endothelial Cells by Imaging Flow Cytometry
 Assessment of Granulocyte Subset Activation: New Information from Image-Based Flow Cytometry
Assessment of Granulocyte Subset Activation: New Information from Image-Based Flow Cytometry
 Accurate Assessment of Cell Death by Imaging Flow Cytometry
Accurate Assessment of Cell Death by Imaging Flow Cytometry
 Screening for Drugs Against the Plasmodium falciparum Digestive Vacuole by Imaging Flow Cytometry
Screening for Drugs Against the Plasmodium falciparum Digestive Vacuole by Imaging Flow Cytometry




Optimising the PI concentration for IFC analysis (a) Fixed Jurkat cells were stained with decreasing levels of PI and acquired at increasing 488 nm laser powers as indicated. The PI fluorescence was analyzed by exporting as an FSC file and using the modeling option in FlowJo [33]. In all cases the G1 CV was <9 and the % of cells modelled in each phase was comparable (data not shown). (b) Fixed Jurkat cells were labeled with 50 μg/ml PI and acquired on a 12-channel IFC system using the indicated 488/405 nm laser powers. The 45° lines drawn through the origin for each plot denote the limits of acceptable spill over whereby PI fluoresce becomes brighter in CH11 than CH5 and would therefore require over 100 % compensation (not possible by IFC)
Related posts:
Quantitative Functional Morphology by Imaging Flow Cytometry
Principles of Amnis Imaging Flow Cytometry
Detection and Characterization of Rare Circulating Endothelial Cells by Imaging Flow Cytometry
Assessment of Granulocyte Subset Activation: New Information from Image-Based Flow Cytometry
Accurate Assessment of Cell Death by Imaging Flow Cytometry
Screening for Drugs Against the Plasmodium falciparum Digestive Vacuole by Imaging Flow Cytometry
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree