Fig. 11.1
Pediatric CNS tumors according to histology
11.2.2 Clinical Presentation
11.2.2.1 Initial Signs and Symptoms
Initial signs and symptoms depend on the primary site of the neoplasm in relation to the cerebral structures and on the child’s age [114].
11.2.2.2 Signs of Endocranial Hypertension
Signs of endocranial hypertension can develop quite late, partly because of the skull’s plasticity (which is all the greater the younger the patient involved) and partly because the brain is still malleable in childhood and may be compressed without any neurological changes becoming apparent.
11.2.2.3 Ocular Fundus Anomalies
In the newborn, the fundus may be normal despite endocranial hypertension, whereas older children more frequently show signs of papillary edema and hemorrhage, sometimes associated with a decline in visual acuity and even amaurosis. It is worth bearing in mind that papillary edema is always a sign of endocranial hypertension, but the absence of the former does not rule out the latter.
11.2.2.4 Signs of Brain Herniation
If endocranial hypertension goes undiagnosed, there may be signs of brain herniation:
Uncal (tentorial) herniation: due to craniocaudal pressure sufficient to make the deepest and most medial part of the temporal lobe (uncus) herniate through the tentorial notch, compressing the brainstem structures, interfering with the state of vigilance, and causing motor pathway suffering with hemiparesis–hemiplegia of the contralateral side and third cranial nerve impairment
Cerebellar (subtentorial) herniation: engagement of the cerebellar tonsils in the occipital foramen, reaching as far as the bulb and spinal canal and causing an altered state of consciousness, bradypnea and bradycardia, episodes of decerebrate and opisthotonic posturing, and stiffness.
11.2.2.5 Focal Signs
Supratentorial tumors may be disclosed by lateral impairment syndromes, partial epileptic seizures, psychic changes (mood disorders, abulia, poor academic performance), and eyesight impairments, depending on the suprachiasmatic sites involved. In tumors of the posterior fossa, there may be evidence of cerebellar syndrome and/or progressive hemiplegia associated with eye movement paralysis. There may also be concomitant neuroendocrine disorders, such as a late or early onset of puberty, delayed height or weight gain, and trophic alterations in the case of diencephalic neoplasms (diencephalic syndrome becomes manifest with failure to thrive). Optic nerve gliomas can become apparent from a decline in visual acuity and proptosis.
11.2.3 Diagnostic Workup: Instrumental Diagnostics and Staging
When clinical signs and symptoms are suggestive of CNS tumors, it becomes mandatory to conduct specific instrumental investigations because no blood chemistry tests are capable of orienting toward a diagnosis of neoplastic disease [82].
1.
Computerized axial tomography (CT): This method is useful in emergency conditions to assess hydrocephalus, bleeding, and liquor or blood collections or to identify calcified lesions.
2.
Nuclear magnetic resonance imaging (NMRI): This is the standard method for diagnosing patients with a suspected CNS neoplasm and for assessing response to treatment. MRI provides additional anatomical details concerning the tumor’s topography, extent, and solid and liquid components. Nuclear MRI enables images to be constructed on sagittal, coronal, and axial planes that are particularly useful for assessing the anatomy of the posterior fossa and brainstem. T1 sequences provide a good degree of anatomical detail, while differences between normal and pathological tissues emerge on T2 images. Imaging with fluid attenuation inversion recovery (FLAIR) and T2 sequences reveal areas where the blood–brain barrier is disrupted and evidence of neoplastic infiltration and edema. NMRI can document areas of treatment-related leukomalacia so it has an indispensable role in the follow-up of iatrogenic sequelae too.
3.
Positron emission tomography (PET): This method may be useful for distinguishing tumor recurrence from radiation-induced necrosis, especially in tumors with a high level of metabolic activity. Malignant tumors tend to be hypermetabolic compared with surrounding tissues, while necrotic tissue is hypometabolic. This distinction is scarcely applicable to childhood brain tumors, however. PET can also be used to highlight functionally important and therefore unresectable brain areas in children. A chapter in this book is dedicated to the characteristics and diagnostic value of PET.
The purpose of staging is to establish the extent of the tumor, its dimensions, and any subarachnoid dissemination. Extracerebral and lymph node metastases are extremely rare.
Staging relies on the following methods:
1.
Postoperative NMRI of the brain and spinal cord, with and without contrast medium: The spinal cord should preferably be assessed before surgery to avoid false-positive results. Imaging within 48 h after surgery can provide a reliable idea of residual tumor volume (which may have prognostic implications and may be used to decide for the therapeutic strategy).
2.
Diagnostic lumbar puncture: This test is indispensable for identifying any liquoral dissemination of the disease (liquor cytology) or liquor circulation dysfunctions (biochemical tests). It can also be used to test for specific markers (such as αFP and βHCG, which are diagnostic of germ cell tumors).
No unified staging system currently exists for CNS tumors.
11.2.4 Pathology
The currently used histological classification was developed by the World Health Organization (WHO) and was last revised in 2007 [63]. In recent years, molecular biology and molecular cytogenetics have gained importance for identifying tumor-specific genetic changes and molecular markers of prognostic and/or therapeutic interest, influencing the histopathological classification of these neoplasms. In the biological setting, the most important recent discovery concerns the isolation, in pediatric as well as adult brain tumors, of variable proportions of genetically modified cells (tumor stem cells) with the distinctive characteristics of specific neural precursors existing at various brain sites, i.e., the expression of specific immunophenotypic markers and their expression profiles relating to signal pathways involved in neural development, a capacity for self-renewal, and a capacity for generating a variety of tumor cells. The main histopathological and molecular characteristics of the tumors most often encountered in pediatric age – i.e., gliomas, neural neoplasms, embryonic neoplasms, germ cell tumors, choroid plexus tumors, and meningiomas – are listed below.
11.2.4.1 Gliomas
Gliomas derive from glial cells and include astrocytomas, oligodendrogliomas, ependymomas, and choroid plexus tumors.
Pilocytic astrocytoma (WHO grade I) originates mainly in the cerebellum but also in the third ventricle, the optic and chiasmatic pathways, the basal ganglia, and the spinal cord. B-RAF gene mutations are quite common, being found in 20 % of these tumors in pediatric age. Diffuse astrocytoma (WHO grade II) usually involves the brainstem and spinal cord. Anaplastic astrocytoma (WHO grade III) and glioblastoma (WHO grade IV) are morphologically similar to their counterpart in adults, but in children, the p53 mutation is more common, while EGFR, PTEN, p14, and p16 gene mutations are rare. Glioblastoma multiforme (grade IV) is an astrocytic variant in which the anaplastic features dominate the picture. Malignant gliomas account for approximately 10–15 % of brain tumors in children.
Ependymomas (10 % of pediatric brain tumors) derive from the ependyma and develop in the vicinity of the ventricular structures, including the spinal canal. In children and young adults, they tend to occur at intracranial sites, with a predilection for the fourth ventricle. In adolescents and older adults, spinal localizations are more common [37].
Most ependymomas are classified as WHO grade II. Less frequently, they may show histological signs of anaplasia with numerous mitosis, vascular proliferation and necrosis, and a more aggressive clinical behavior (anaplastic ependymoma, WHO grade III).
NF2 gene mutations are found in cases of spinal ependymoma, but not in patients with intracranial tumors. Gain of 1q has been associated with a worse prognosis. Gene expression analyses have revealed the activation of various pathways involved in neural development and NOTCH in particular [76, 95].
Choroid plexus tumors derive from the specialized choroid plexus cells responsible for producing the cerebrospinal fluid. In pediatric age, they occur mainly within the lateral ventricles.
11.2.4.2 Neural and Mixed Glioneuronal Neoplasms
Neural and mixed glioneuronal neoplasms are rare and generally have a favorable course. They tend to occur mainly in adolescents and young adults. The sites most often affected are the temporal lobe, the floor of the third ventricle, and the hypothalamus. Histologically, these neoplasms consist of mature neural cells (gangliocytoma) sometimes associated with a glial component (ganglioglioma). This group also includes even rarer forms, such as neurocytoma, dysembryoplastic neuroepithelial tumor (DNET), and glioneuronal papillary tumor. These lesions often cause long-term drug-resistant epilepsy. Desmoplastic infantile astrocytoma/ganglioglioma (WHO grade I) is a separate entity.
11.2.4.3 Embryonal Neoplasms
Embryonal neoplasms derive from primitive embryonal cells. They typically develop early in childhood and are highly malignant, and consequently all are classified as WHO grade IV. Embryonal neoplasms include medulloblastoma, primitive neuroectodermal tumors (PNETs), and atypical teratoid/rhabdoid tumor (AT/RT).
Medulloblastomas account for 10–20 % of BTs and 40 % of tumors of the posterior fossa, often arising from the cerebellar vermis or fourth ventricle. Various subtypes (classic, nodular/desmoplastic, large cell/anaplastic) have been identified that correlate with prognosis and are consequently now used to orient the choice of treatment. Other molecular subtypes and genetic patterns of prognostic significance have emerged from recent studies, i.e., ERB-B2, TRKC, PDGFRA, MYCC, and MYCN expression of β-catenin, and will be considered in upcoming clinical trials. Seeding may occur into the subarachnoid space, CSF, or supratentorial sites and rarely into the bone marrow, bones, and liver [68, 97].
Non–cerebellar PNETs account for 2–3 % of pediatric CNS tumors. They are generally localized in one of the hemispheres and are locally extensive, often with leptomeningeal dissemination.
11.2.4.4 Germ Cell Tumors
Germ cell tumors make up 3.5 % of pediatric brain neoplasms, with a higher incidence in males and a mean age of onset around 12 years. The sites most often affected are located along the midline, in the pineal gland, the suprasellar region, and the third ventricle [79]. At CNS level, a distinction is drawn between secreting and nonsecreting germinomas, depending on the presence of pathological levels of αFP and/or βHCG in the serum and/or liquor.
11.2.4.5 Meningiomas
Meningiomas develop mainly in adult age, representing almost 20 % of all intracranial primary neoplasms. They may occasionally develop in pediatric age too. The most common molecular change involves the loss of chromosome 22 with NF2 gene deletion or mutations. There is a high incidence of meningioma, often involving multiple sites, in patients with neurofibromatosis type II, in which the gene is mutated at germinal level.
11.2.5 Treatment and Follow-Up
This always includes primary surgery, whenever it is technically feasible. Complementary treatment with chemotherapy and/or radiotherapy may be provided, depending on the outcome of surgery and on the histotype involved. In all cases, a patient’s treatment should be planned jointly by a team including the neurosurgeon, pediatric oncologist, pathologist, radiotherapist, and neuroradiologist.
11.2.5.1 Surgery
Indications
Surgery is indicated in the majority of cases, and children should ideally be operated only by surgeons specializing in pediatric diseases.
Treatment for Hydrocephalus
The indication for surgery must take into account any hydrocephalus, which should always be treated before any attempt at surgery on the tumor mass. Hydrocephalus can be treated by means of shunts (internal or external ventricular shunts, depending on the duration of the drainage) or endoscopically [45].
11.2.5.2 Radiotherapy
Irradiation has a role in preventing local recurrences and tumor spread. When an adjuvant chemotherapy is used before radiotherapy, it is important to bear in mind that some chemotherapeutic agents have a toxicity of their own that compounds the expected toxicity of the radiation treatment (for instance, methotrexate is neurotoxic and cisplatin can sensitize the inner ear to radiation-induced damage). Depending on the tumor’s histology, the volume to irradiate may coincide with the tumor alone (administering 50–55 Gy) or the tumor plus the neural axis (administering from 23 to 36 Gy on a level with the brain and spinal cord). Radiotherapy is administered in daily fractions of 1.6–1.8 Gy for 5 days a week. Hyperfractionation (administering more than one fraction a day in reduced doses, e.g., 1.1–1.3 Gy every 6–8 h) is an unconventional method used in controlled clinical trials with a view of increasing the biologically effective dose of radiation while reducing the side effects on the healthy tissues.
All candidates for radiotherapy to the CNS should be treated after formulating a treatment plan with the aid of software capable of generating 3D images from combinations of MRI and CT findings. The term “conformal radiotherapy” refers to any radiotherapeutic method that enables the distribution of the therapeutic dose to be shaped to fit the target tumor volume, reducing the level of radiation absorbed by the surrounding healthy tissues. This can be achieved using various methods, such as fixed-beam 3D conformal radiotherapy, multiple-arc stereotactic radiotherapy, intensity-modulated radiation therapy (IMRT), tomotherapy, and so on. Another approach exploits the peculiar physical characteristics of hadrons, and of proton beams in particular, to make the radiation even more conformable and thus better safeguard adjacent healthy tissues [72].
11.2.5.3 Chemotherapy
The presence of the blood–brain barrier makes chemotherapy less effective in BTs than in solid tumors occurring at other sites. The drugs most often used to treat pediatric brain neoplasms are the nitrosoureas, vincristine, procarbazine, platinum derivatives, epipodophyllotoxins, high-dose methotrexate, and other antimetabolites. Among the drugs used most recently, it is worth mentioning temozolomide, which achieves a good bioavailability after oral administration and causes minimal myelosuppression.
Intrathecal and intraventricular chemotherapy relies mainly on methotrexate and should be omitted after radiotherapy to the neural axis to avoid cumulative toxicity phenomena. Some protocols recommend chemotherapy before radiotherapy, as early as possible after surgery, based on the assumption that postoperative anatomical changes may facilitate the drugs’ passage through the blood–brain barrier. One way to overcome the obstacle represented by the blood–brain barrier consists in depositing the drug directly within the tumor bed. In selected cases, and subject to the expertise available at the center involved, high-dose chemotherapy followed by autologous marrow or peripheral stem cell transplantation may be used postoperatively for neoplasms with an unfavorable prognosis relating to their histotype or stage or to the patient’s age. This treatment suffers from a high morbidity rate and high costs, however, and only centers specializing in pediatric oncology and equipped to perform this type of treatment can adopt such complex therapeutic protocols.
11.2.5.4 Biological Treatment
The characterization of new signal transduction pathways implicated in the genesis and progression of CNS tumors has given rise to a number of new drugs and therapeutic approaches for neoplasms that fail to respond to conventional treatments. Such novel agents include specific inhibitors (small molecules), monoclonal antibodies (moAb), and angiogenesis inhibitors. The majority of these new drugs need further clinical testing (some of which is currently under way) to establish their optimal dosage, alone or in combination, and to ascertain their additive potential or possible synergic effects.
Using vaccines obtained by charging dendritic cells with glioblastoma proteins or tumor lysates, various phase 1 studies have demonstrated that vaccination is a safe procedure and capable of contributing to a significant improvement in overall survival. Research in pediatric neuro-oncology has tested the feasibility of this procedure in children too, particularly in cases in which recurrence have been macroscopically completely resected. It remains to be seen whether such cell therapy approaches can be included in multicenter protocols, given the assurance of good clinical practice that such a refined cell manipulation demands.
11.2.5.5 Salvage Therapy
It is not always advisable to attempt further therapies if a brain tumor evolves despite primary treatment. A second surgical procedure should always be considered, albeit with the limitations intrinsic in brain anatomy and the greater risks associated with resurgery in tissues that have become abnormally revascularized after radiotherapy – especially in neoplasms of low-grade malignancy and ependymomas. Where feasible, radiotherapy could be repeated, in doses depending on the site of the recurrence and the dosage already administered. Various other drugs, from the classic chemotherapeutic agents to the biological options, alone or in association, have also been used as second-line treatments.
Flowchart 11.1 shows the general diagnostic workup for CNS neoplasms. Then the last part of this chapter goes into further detail concerning the diagnosis and treatment of the main histotypes.
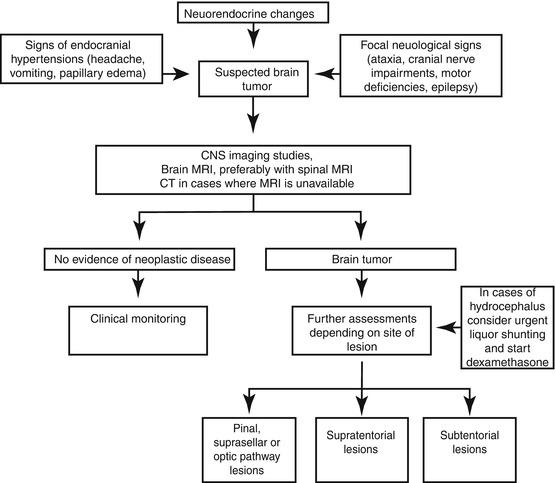
Flowchart 11.1
Diagnostic flow-chart of brain tumors
Oncological Follow-Up
This involves regular clinical and radiological assessments, their frequency depending on the type of tumor, the treatment administered, and the time elapsing since its completion [81]. Children with severe neurological impairments immediately after surgery are unlikely to recover completely, and persistently high intracranial pressures can lead to atrophy of the optic pathways and visual impairments. Children presenting with supratentorial disease may have convulsions even after completing their treatment and consequently require long-term antiepileptic medication.
In addition to follow-up visits, objective neurological assessments and NMRI, neuropsychological and endocrinological assessments should be an integral part of the follow-up for children treated for CNS neoplasms. Nowadays, more than one in two children who develop brain tumors have a chance of being cured and reaching adulthood, but the price to pay is often high in terms of the sequelae, which become manifest in neurocognitive, endocrinological–metabolic, and somatic growth impairments. Efforts focusing on the prevention, rehabilitation, and correction of these deficits are consequently an integral part of the treatment plan for children with brain tumors.
11.2.6 Gliomas
11.2.6.1 Low-Grade Histotypes
Maximally safe resection (generally achievable for hemispheric or cerebellar locations) has significant prognostic implications in these tumors. They are typically well circumscribed but quite frequently include a subtle component that may be hard to differentiate from normal surrounding brain tissue. Real-time imaging with navigational capabilities may be helpful during surgery to maximize resection control. Chemotherapy is not generally necessary, but several clinical trials have demonstrated that it has a role in stabilizing or shrinking low-grade gliomas, so it is used in progressive or unresectable tumors. The most widely used chemotherapy regimens are carboplatin and vincristine, cisplatin and etoposide, and vinblastine [80]. Radiotherapy is not a first choice (due to its long-term side effects in populations with an excellent prognosis). In fact, radiation-induced tumors are a concern in patients with low-grade glioma associated with genetic syndromes, such as phacomatosis. These conditions carry a very good prognosis, with 5-year overall survival rates > 90 % after complete (and often curative) surgical resection. The follow-up should be more frequent in the event of residual disease, with or without ophthalmological assessment for optic pathway gliomas, and/or endocrinological assessment, depending on the site of the lesion.
11.2.6.2 High-Grade Histotypes
These neoplasms have an infiltrative growth pattern and a rapid growth rate and often recur after treatment. At present, the strongest prognostic indicator is the extent of resection, but radical surgery is often not feasible due to the infiltrative pattern and the tumor’s location, frequently involving the thalamus and pons. Biopsy is advisable in cases of diffuse intrinsic pontine glioma (DIPG) or gliomatosis cerebri to ascertain the disease’s biological characteristics and orient the choice and targeting of treatments, especially in clinical trials. High-dose focal radiation after maximal surgery is the best treatment strategy. There is currently no officially acknowledged standard chemotherapy, but, based on experience in adults, adding oral temozolomide to radiotherapy and for maintenance has long been considered as such [109]. Myeloablative chemotherapy is not standard but is used for first-diagnosed and relapsing tumors, with variable results [65]. Target therapies (gefitinib, erlotinib, imatinib, nimotuzumab) are currently being tested in combination with conventional therapies [64]. The median overall survival for patients with high-grade tumors is around 12–24 months.
11.2.7 Medulloblastoma (Cerebellar PNET)
Maximal safe resection is the mainstay of treatment. Postoperative staging enables patients to be classified as standard risk (residual disease ≤1.5 cm2, no metastases) or high risk (residual disease >1.5 cm2 and/or metastases). Chemo- and radiotherapy (RT) are both mandatory after surgery. The aim of RT is to control residual disease and avoid subarachnoid dissemination. In cases of localized disease, the standard treatment includes craniospinal irradiation (CSI; 23.4 Gy) plus a boost on the tumor site (30.6 Gy) and chemotherapy using various schedules (vincristine, cisplatin, etoposide, carboplatin, cyclophosphamide, and lomustine). For metastatic disease, the strategy currently used at our institute includes pre-radiation intensive chemotherapy and hyperfractionated accelerated radiotherapy (HART), with or without subsequent consolidation with myeloablative chemotherapy. With this treatment approach, the event-free survival rate (EFS) at 5 years rises from 50 to 70 % [43].
11.2.8 PNETs: Pineoblastoma
After maximal safe resection, adjuvant therapy consists of radiation to the tumor bed (40–60 Gy) and, in selected cases, to the neuraxis (30–35 Gy), associated with chemotherapy (nitrosoureas, procarbazine, cyclophosphamide, vincristine, cisplatin, and carboplatin). This approach has recently been intensified with the addition of myeloablative schedules in order to reduce the radiation doses/fields and improve the patient’s prognosis [34]. Only 35 % of patients are alive at 5 years.
11.2.9 Ependymoma
Gross-total safe resection has a primary prognostic influence: The 5-year survival rate is about 70 % for near complete resections as opposed to about 40 % for subtotal resections. Second-look surgery is justified when postoperative imaging suggests residual disease or during treatment. Radiotherapy is essential, even after complete resection, with total doses to the tumor bed of 59.4 Gy; the role of a boost to any residual disease is the object of ongoing international studies [66]. Adjuvant chemotherapy can be recommended, within controlled clinical trials, in cases of residual disease or unfavorable histology to shrink the residual tumor and enable total resection at second-look surgery [69]. Late recurrences can occur, for which complete surgery is always the goal, possibly followed by re-irradiation.
11.2.10 Germ Cell Tumors (GCT)
There are two subtypes of this disease: pure germinomas (accounting for two in every three GCTs) and “nongerminomatous germ cell tumors” (NGGCT), which can be associated with pathological αFP/βhCG findings in the serum or CSF. Surgical removal is not recommended because of these neoplasms’ high chemo- and/or radiosensitivity, but surgery is important in establishing the histological diagnosis as well as for the treatment of hydrocephalus, and it is essential for diagnostic purposes if αFP/βhCG levels in the serum or CSF are not pathological. Debulking procedures may be recommended in NGGCT as they are often chemotherapy resistant. To contain or avoid exposure to radiation, platinum-based chemotherapeutic regimens are used to good effect. Radiotherapy fields should include whole ventricular systems. RT alone on the whole CNS can be curative in cases of metastatic disease. Germinomas have long-term survival rates >90 %, while NGGCTs carry a worse prognosis, with a 5-year overall survival (OS) rate of 60–70 %, and treatment for the latter must include both platinum-based regimens and radiation therapy. Endocrinological sequelae should be carefully monitored during the follow-up and also the markers found pathological at diagnosis [56].
11.2.11 Brainstem Tumors
These are usually astrocytomas; they account for 10–15 % of all pediatric intracranial tumors and 20 % of posterior fossa tumors. They have different degrees of malignancy and infiltrate the brainstem extensively. Symptoms are usually rapidly progressive.
Among the three patterns seen in brainstem lesions, diffuse infiltrating pontine glioma (DIPG) is the most common. A single cycle lasting 5–7 weeks of radiotherapy (54 Gy) is generally recommended, but the median OS is less than 1 year.
Neither standard nor high-dose adjuvant chemotherapy has proved capable of modifying the prognosis. Recent molecular studies have shed light on the genetic profiles of DIPG, showing that EGFR, PDGFR, and H3F3D are involved in different subsets of these gliomas [112, 115]. These findings may have great promise when applied to new therapeutic strategies in the future.
11.2.12 Atypical Teratoid/Rhabdoid Tumor (AT/RT)
This is an aggressive neoplasm characteristic of infancy with a dismal prognosis [21]. It is usually located in the posterior fossa and cerebral hemispheres. It has been recognized as a separate entity from PNET/medulloblastoma due to the presence of rhabdoid cells and components of malignant mesenchymal and epithelial cells. It is cytogenetically characterized by monosomy of 22q11 (and the hSNF/INI1 gene is involved).
A sizable proportion of cases carries de novo SMARCB1 constitutional mutations in the setting of the “rhabdoid tumor predisposition syndrome,” and the outcome is worst in infants with syndromic AT/RT. Multiple therapeutic approaches have been attempted over the last two decades to improve survival, without success. There is no accepted standard chemotherapy, but intensive alkylator-based chemotherapy, high-dose methotrexate, and high-dose regimens with stem cell rescue may prove more effective. Focal radiation is a crucial treatment. Overall survival is less than 30 % at 12 months, and progression-free survival (PFS) at 1 year is less than 20 %.
Table 11.1 shows the survival rates for the various types of CNS neoplasm using the currently recommended treatments [10].
Table 11.1
Treatment and prognosis of most frequent pediatric brain tumors
Tumor | Therapy | Overall survival % at 5 years |
|---|---|---|
Gliomas | ||
Cerebellar astrocytoma | ||
Low-grade, complete surgery | S | 90 |
High-grade, partial surgery | S, LR ± C | 15 |
Diffuse pontine glioma | LR ± S (±C) | <10 |
Supratentorial astrocytoma | ||
Low-grade: | ||
Complete surgery | S | 80 |
Partial surgery | S ± LR (±C) | 70 |
Ηigh-grade | S, LR, C | 35 |
Glioblastoma | S, LR, C | 10 |
Medulloblastoma | ||
Age ≥ 3 years | S, LR, CSI, C | 80 |
Age < 3 years | S, C, ± LR/CSI | 70 |
Ependymoma | ||
Complete surgery | S, LR | 70 |
Partial surgery | S, LR ± C | 40 |
Pineal region | ||
Germ cell tumors (all) | S, LR ± CSI ± C | 70 |
Pineoblastoma | S, CSI, LR ± C | 40 |
Other non-cerebellar PNETs | S, CSI, LR ± C | 40 |
Choroid plexus carcinoma | S, LR ± C | 40 |
All tumors | 60 |
11.3 Lymphomas
11.3.1 Epidemiology
Non-Hodgkin Lymphoma
Non-Hodgkin lymphoma (NHL) accounts for approximately 10–15 % of all malignant neoplasms of pediatric age, and their incidence rises constantly with age. The etiology of NHL remains largely unknown, with the exception of significant associations with Epstein–Barr viral infection and African Burkitt lymphoma and a predisposition associated with certain congenital syndromes related to immunodeficiency. Overall, in Europe and the United States, 50–60 % of cases of pediatric NHL are Burkitt/Burkitt-like and mature large B-cell NHL, while 30 % are lymphoblastic lymphomas (mainly intermediate or late precursor T-cell and, less frequently, B-cell neoplasms), and 10–15 % are anaplastic large cell lymphomas. Other variants that are common in adults are rare in children and adolescents.
Hodgkin Lymphoma
Hodgkin lymphoma (HL) accounts for approximately 6 % of pediatric neoplasms. It shows a predilection for males, especially in younger age, and a bimodal distribution in its incidence, with a first peak between 15 and 30 years of age and a second beyond the age of 50. For HL too, there is no clear etiology, with the exception of a frequent association with high anti-EBV antibody levels and with EBV genomes in the Hodgkin and Reed–Sternberg cells, particularly in developing countries.
11.3.2 Clinical Presentation
The clinical presentation depends on the histotype, but it always correlates with the site affected by the disease. Often it is associated with systemic symptoms such as fever, weight loss, swallowing, pruritus, and asthenia.
11.3.3 Diagnosis and Staging
Staging
Cytohistology is a fundamental element in the diagnosis of lymphoma. When adenopathies are present, lymph node biopsy and lymphadenectomy are mandatory (whereas fine needle biopsies should be avoided because of their limited diagnostic value). If there is evidence of ascitic fluid or pleural effusion, cytologic examination may substitute histology for diagnosis. In addition to the standard radiological investigations (CT/MRI) of the chest and abdomen and of the affected region, staging includes bilateral bone marrow evaluation and diagnostic lumbar puncture for NHL, while only bone marrow biopsy is indicated for HL. Some authors have reported that FDG-PET can accurately predict the outcome of bone marrow biopsy for staging purposes, suggesting that bone marrow biopsy may be omitted in patients with PET findings of bone marrow involvement [18]. The staging of pediatric NHL is that developed by Dr. Murphy at the St. Jude Children’s Research Hospital in Memphis, while HL is currently staged using the Ann Arbor protocol (1971), as revised in 1989.
The role of PET
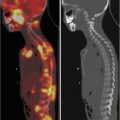
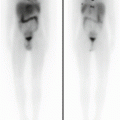
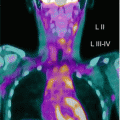
Over the last decade, PET has gradually replaced gallium scan as the preferred functional imaging modality for lymphoma staging. In adult lymphomas, several studies have shown the superiority of PET/CT over classic radiological investigations for the initial workup and for assessing remission. The ability of PET/CT imaging to integrate the tumor’s functional and anatomical characteristics has prompted its use for staging and monitoring pediatric patients with lymphoma because it is both accurate and cost-effective. Emerging studies also tend to support the prognostic value of a persistent PET/CT positivity after 2 chemotherapy courses, which could warrant an early assessment of the refractory disease. Very limited data are available on the specificity and sensitivity of this method in childhood NHL (apart from primary mediastinal large B-cell lymphoma, which is similar in children and adults), but some studies report high sensitivity and specificity rates for FDG-PET in assessing treatment response. The ability of FDG-PET to image hypermetabolic tumors could make it a reliable method for detecting all sites of disease at diagnosis. Information from FDG-PET may influence treatment planning by prompting an upstaging or downstaging of the disease. It may therefore spare downstaged patients the long-term side effects of intense chemotherapy and enable more effective treatment to be offered for upstaged lymphomas. In contemporary, risk-adapted chemoradiotherapy regimens, response to therapy is frequently a criterion for tailoring therapy. Identifying the metabolic tumor activity through PET serves as a surrogate measure of tumor response and often makes it unnecessary to document disease status with invasive procedures.
In the pediatric population, PET imaging has some peculiarities: To obtain an optimal scan, the patient must lie quietly to minimize FDG uptake related to muscle movements and not to tumor activity and also to minimize the need of a sedation. Even in ideal conditions, the avidity of FDG for certain structures such as brown fat or the cervical musculature may hinder the interpretation of signs of nodal involvement by lymphoma. An abnormal FDG avidity may be seen in patients treated with granulocyte growth factors (involving a transient, intense FDG avidity in the spine, pelvis, and spleen) or in images of patients recovering from an infectious/inflammatory process. During the follow-up, PET can be useful only when standard radiological methods pose a doubt of relapse. Clinicians should be aware that – in certain conditions – PET findings may mimic relapse in the off-therapy setting: Thymic rebound hyperplasia characteristically appears in the 6–12 months after completing the treatment and is characterized by FDG avidity. Prospective studies on PET in pediatric patients will improve our understanding of the optimal use of this method and the characteristics of other nonmalignant conditions that may be considered in the differential diagnosis.
11.3.4 Pathology
The histopathological and immunohistochemical evaluations are fundamental for the diagnosis of NHL and HL, but they should be completed with the genetic and molecular profiling of each histotype. The currently used histopathological classification is based on the latest version of the WHO classification of lymphoproliferative diseases.
Non-Hodgkin Lymphoma
The main histotypes are mature B-cell NHL, precursor T-cell or B-cell NHL, and anaplastic large cell NHL
B-cell NHL: These comprise Burkitt lymphoma and diffuse large B-cell lymphoma and primary mediastinal large B-cell NHL which is typical in adolescent females.
Burkitt lymphoma has a morphological appearance of a “starry sky” due to histiocytes and macrophages coexisting among the blasts. Cytologically, this disease consists of type L3 cells according to the FAB classification, with a very high mitotic index (>90 %). The cells express surface immunoglobulins (Ig) and are positive for several markers, including CD19, CD20, CD22, CD79a, and often CD10. TdT is negative.
Large B-cell lymphoma expresses a similar phenotype, but CD10 may be negative, and some patients do not express surface Ig. Mediastinal large B-cell NHL is frequently characterized by sclerosis, and it is often CD30 positive so that differential diagnosis with HL is possible.
Lymphoblastic lymphoma (LBL): It has a cytological picture similar to lymphoblastic leukemia: the cells have a high nucleus/cytoplasm ratio and finely dispersed chromatin. The most relevant feature for diagnostic purposes is TdT expression, since no more than 5 % of LBLs are TdT negative. The majority of LBLs are also CD99 positive. Approximately 80 % of LBLs derive from T cells and variably express markers that can be traced back to thymic ontogenesis, including CD7, CD2, CD5, CD1, CD3, CD4, and CD8. Precursor B-cell LBLs express such markers as CD19, HLA-DR with CD10, but not surface Ig.
Anaplastic large cell lymphoma (ALCL): It includes several morphological variants, although the so-called common type accounts for approximately 70 % of cases. Differential diagnosis with HL is sometimes difficult. Typically, ALCL is positive for CD30, EMA, T markers, and very often ALK, while it is negative for CD15.
Hodgkin Lymphoma
It includes:
Classic HL (nodular sclerosis, mixed cellularity, lymphocyte depleted, lymphocyte rich)
Nodular lymphocyte-predominant HL
The typical cell of HL is the Reed–Sternberg (RS) cell, which is binucleated or multinucleated, with evident nucleoli immersed in a reactive type of cell matrix (lymphocytes, granulocytes, eosinophils, macrophages). Hodgkin cells are almost always CD30 positive, and they frequently express CD15; they are negative for T-cell markers but sometimes positive (in approximately 20 % of cases) for CD20. The most common form is nodular sclerosis subtype.
11.3.5 Treatment and Follow-Up
NHL
The treatment of NHL is based essentially on polychemotherapy. The role of surgery is generally limited to the diagnostic phase (biopsy) or for assessing residual tumor. Radiotherapy (RT) is not used in the frontline treatment. The use of prophylactic irradiation of the central nervous system for lymphoblastic lymphoma has been abandoned.
The treatment must be designed to obtain a rapid and complete remission and can be summarized in three phases:
Induction phase | Polychemotherapy comprising cyclophosphamide, vincristine, adriamycin asparaginase, and corticosteroids, aiming for a complete remission in 4–6 weeks |
Consolidation phase | Use of high-dose methotrexate and/or cytosine arabinoside to consolidate the complete remission |
Maintenance phase | Cyclic administration of various antimetabolites |
HL
Treatment is based on a combination of chemotherapy and RT, according to the stage of the disease. Efforts are currently being made to reduce the dose of RT in order to contain late effects. Because of the improvements in the prognosis, more attention has to be paid to the toxicity of combinations of chemo- and radiotherapy: The goal of undergoing studies is to maintain a high event-free survival while reducing the use of alkylating drugs and the total dose of RT. As a part of the follow-up, iatrogenic sequelae should be investigated, i.e., hormonal status, organ function (lung, heart, kidney, liver), fertility, and hematological disorders.
11.4 Soft Tissue Sarcomas: Rhabdomyosarcoma
11.4.1 Epidemiology and Etiology
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in pediatric age. It accounts for approximately 4–5 % of all childhood tumors, with approximately 200–250 new cases a year in Italy. It is a highly aggressive neoplasm, comprising cells of mesenchymal origin differentiating into striated muscle. Its incidence peaks at around 2–3 years of age, with a second peak around 15–19 years old. The male gender is slightly more affected [88]. There is a known association with congenital malformations, particularly when they involve the genitourinary tract, and with other conditions such as the Li–Fraumeni and Beckwith–Wiedemann syndromes and neurofibromatosis type 1 [5].
11.4.2 Clinical Presentation
RMS may potentially occur anywhere in the body, but the sites most often affected are the head and neck (40 %), the genitourinary tract (20 %), and the limbs (20 %). The site is an important prognostic factor and influences the therapeutic strategy.
In approximately 20 % of cases, there are already metastases by the time RMS is diagnosed. The disease metastasizes mainly to the lungs and less frequently to the skeleton and bone marrow. There are lymph nodes involved in 20 % of cases, particularly in tumors located in the retroperitoneum, lower limbs, and genitourinary tract.
11.4.3 Diagnostic Workup and Staging
Histological examination (on open or Tru-Cut biopsy material) is essential to the diagnosis.
Local staging demands the use of axial CT and/or MRI with a contrast agent, depending on the site of origin of the tumor (Fig. 11.1). Chest CT scanning, bone scintigraphy with 99Tc, abdominal ultrasound, and bone marrow aspirates and biopsies are needed to stage remote dissemination. Lumbar puncture is also required in parameningeal sites.
The application of FDG-PET appears to be attracting growing interest, but the method is still under study in this setting. FDG-PET may be particularly useful for the staging and reassessment of RMS occurring at certain sites, such as the parameningeal, that are often not amenable to surgery. In such cases, using FDG-PET in combination with MRI may orient the clinician’s actions at the end of the frontline treatment phase, also in relation to the presence or absence of any significant metabolic uptake [89].
The longest-standing staging systems are the ones proposed in 1972 by the North American Intergroup Rhabdomyosarcoma Study Group (IRSG, for postoperative staging) and the TNM (for clinical–radiological staging) [48]. The latest staging systems also consider relevant prognostic parameters, such as histology, the site of onset of the disease, and the patient’s age.
The site of the disease has an acknowledged role, and RMS developing within the orbit and genital tract or at paratesticular sites have a generally better prognosis, while a worse prognosis is associated with the limbs, parameningeal sites, the bladder and prostate, and “other sites”, such as the trunk. The prognosis is of intermediate severity for RMS of the head and neck (other than the parameningeal) or genitourinary tract (not involving the bladder or prostate). Age also seems to have a prognostic significance, with patients under 10 years old usually having a better prognosis [54].
11.4.4 Pathology
RMS can be divided into two main histotypes: the embryonal (ERMS), which accounts for approximately 75–80 % of all RM, and the alveolar (ARMS), in the other 20–25 % of cases. A botryoid subtype of ERMS develops typically in the hollow organs and especially the vagina, bladder, and nasopharynx. ARMS has a characteristic translocation – t(2;13)(q35;q14) or t(1;13)(p36;q14) – involving the PAX3/PAX5 and FKHR genes responsible for regulating transcription [83].
Immunohistochemical investigations document positivity for proteins regulating muscle differentiation, and particularly cytoplasmic positivity for desmin, and nuclear positivity for MyoD1 and myogenin (Myf4) [87].
The currently used international classification distinguishes between the subtypes on the strength of the morphological features of the disease and their prognostic significance. Botryoid and ERMS have an intermediate/favorable prognosis, while the alveolar histotype has a worse prognosis.
11.4.5 Treatment and Follow-Up
The coordinated use of surgery, chemotherapy, and radiotherapy has gradually led to an improvement in the chances of survival for patients suffering from localized RMS, and nowadays, 70 % of patients are candidates for cure.
Surgery
The goal of surgery is complete tumor resection with free margins (R = 0) [100]. Whenever this is unfeasible already at the time of diagnosis or when surgery exposes a patient to a high risk or certain damage, surgery may be limited to biopsy alone (open or Tru–Cut) because RMS is a highly chemo- and radiosensitive disease.
Radiotherapy
RMS is a radiosensitive cancer, so radiotherapy has an important role in local disease control. For some sites (parameningeal sites and prostate), it is the only local treatment modality because surgery would inevitably be demolitive. The doses commonly used are around 40–50 Gy [98].
Brachytherapy (using sources of Ir-192 and Cs-137) has a role in cases of RMS located in the genitourinary tract and at cervicofacial sites.
The risk of late side effects, particularly evident in children (growth retardation, skeletal malformations, cognitive impairments, endocrine disorders), needs to be considered, and the so-called total burden of therapy should be borne in mind when establishing the treatment plan [86, 108].
Chemotherapy
Chemotherapy has considerably modified the natural history of RMS, proving highly effective in controlling micro-metastases and enabling significant volume reductions in both the primary tumor and any remote metastases.
The drug combinations currently used (“gold standard”) are vincristine, ifosfamide, and actinomycin D (the IVA protocol) [36] or the American alternative, in which cyclophosphamide is used instead of ifosfamide (the VAC protocol). This approach enables objective responses to be achieved in 75–80 % of cases. The duration of the treatment varies from 6 to 12 months.
Other drugs with a documented activity against RMS are the topoisomerase I inhibitors (topotecan and irinotecan), which have achieved response rates in excess of 40 % in previously untreated patients, and vinorelbine, a semisynthetic vinca alkaloid that has demonstrated a good activity associated with a limited toxicity [16].
After completing their treatment, patients with RMS must be followed up carefully, partly because of the risk of recurrences and partly to monitor any side effects of the treatments they have received.
Recurrences are more likely to occur in the first 3 years after the primary RMS was diagnosed, while they are rare beyond 5 years. They are local recurrences in 60 % of cases.
11.5 Non-Rhabdomyosarcoma Sarcomas (“Non-Rhabdo”)
11.5.1 Epidemiology and Etiology
“Non-rhabdo” soft tissue sarcomas form a heterogeneous group of malignant mesenchymal neoplasms that represent approximately 3–4 % of all tumors occurring in pediatric age and adolescence. The neoplasms in this group differ from one another in biological characteristics and clinical history, and – apart from a few exceptions, such as infantile fibrosarcoma – they are diseases more typical of adults (the so-called adult-type soft tissue sarcomas [35]). The histotype most often encountered is synovial sarcoma (Fig. 11.2), which has a peak incidence between 15 and 30 years of age.
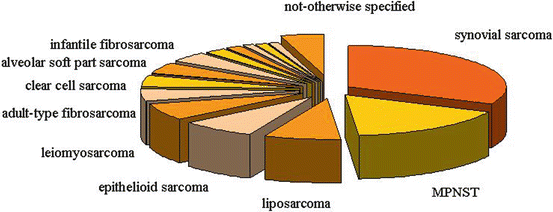
Fig. 11.2
Most frequent histotypes of non-rhabdomyosarcoma sarcomas
11.5.2 Diagnostic Workup and Staging
Although it may develop in virtually any part of the body, the limbs are the most commonly affected site of this tumor.
The disease’s clinical behavior, and particularly its tendency to develop hematogenous metastases (generally to the lung), varies in relation to the histotype and the tumor’s degree of malignancy [106].
The diagnosis can be established on incisional or Tru–Cut biopsies.
Local staging demands MRI with a contrast medium and/or axial CT scanning, while chest CT scans (and sometimes abdominal and brain CT scans for certain histotypes) and possibly bone scintigraphy are used for staging remote disease
11.5.3 Pathology
The histological classification of sarcomas of the soft tissue relies on the line of cellular differentiation, i.e., on a comparison between the tumor’s cell line and the corresponding normal cell line. The histological diagnosis is necessarily based on findings obtained using traditional optical microscopy and immunohistochemistry but also relies increasingly nowadays on site genetics and molecular biology, used to identify chromosomal and molecular rearrangements specific to the various sarcomas.
11.5.4 Treatment and Follow-Up
The treatment of (pediatric or adult) patients with soft tissue sarcoma is rather complex. It demands an integrated, multidisciplinary approach that takes all possible therapeutic options – including surgery, radiotherapy, and chemotherapy – into account.
Surgery
This is the core treatment for this condition. Every effort must be made to ensure that any surgery is radical (with oncologically free surgical margins) and conservative (in both anatomical and functional terms), whenever possible [46].
Radiotherapy
This is indicated for tumors that are unresectable or have undergone marginal surgery, especially in the case of sarcomas of high-grade malignancy or for high-grade (G3) sarcomas greater than 5 cm in diameter that have been widely resected. The role of radiotherapy for sarcomas of low-grade malignancy (which are less sensitive to radiation) is more debatable.
Chemotherapy
While the role of surgery and radiotherapy for soft tissue sarcomas is well established, the part that chemotherapy can play is more uncertain. Systemic treatment is important, however, for high-grade (G3) tumors and those larger than 5 cm in size because they feature a strong tendency to develop remote metastases [41]. A combination of ifosfamide–doxorubicin is the solution most often used. Other drugs (gemcitabine, docetaxel, cisplatin, trabectedin, etoposide) may be used for certain particular histologies.
The methods and timing of the follow-up depend on the risks associated with the degree of malignancy, the tumor’s dimensions, and the radicality of surgery.
Follow-up is always needed to check for possible sequelae of treatments (fibrosis, lymphedema, growth retardation in irradiated regions, endocrine damage, second tumors).
11.6 Bone Sarcomas
Tumors involving the bone form a group of neoplasms with various presenting clinical features, radiological appearances, and histopathological characteristics. They mainly affect patients in the second and third decades of life and essentially consist of osteosarcoma and the Ewing sarcoma family tumors (ESFT).
11.6.1 Osteosarcoma
11.6.1.1 Epidemiology and Etiology
Osteosarcoma (OS) is the most common primary malignancy occurring in the bone, accounting for 60 % of all tumors. It is rare below 5 years of age and above 50 years of age. It has a peak incidence in adolescence, coinciding with the time when the body is growing the most. The annual incidence of OS is 2–3 cases per million population. Males are more affected than females, with a male-to-female ratio of 1.5:1.
Ionizing radiation is considered a risk factor (the risk of an OS developing in an irradiated body region being 2000 times higher than usual). Other risk factors include (1) prior hereditary retinoblastoma (the RB oncogene); (2) Li–Fraumeni syndrome (the p53 oncosuppressor is found mutated in 3–4 % of all cases of OS); and (3) Bloom, Werner, and Rothmund–Thomson syndromes and Paget’s disease (2 % of patients with OS).
11.6.1.2 Clinical Presentation
The clinical features of OS usually include a swelling that rapidly increases in size, with inconstant and increasingly severe pain, and functional impotence, but the presenting sign may sometimes be a pathological fracture. The disease usually develops on a level with the metaphysis of a long bone, and the neoplasm affects the knee (distal femur or proximal tibia) in 75 % of cases.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


