CHAPTER 47 Toxic and Metabolic Brain Disease
Toxic and metabolic brain disease encompasses a vast and heterogeneous group of disorders that can cause a great deal of confusion to both clinicians and radiologists. Diagnosis of these disorders can be challenging, because their clinical and imaging characteristics are often nonspecific. A number of systems have been proposed to classify these disorders and have been based on, among other things, clinical features, histologic features, and biochemical features. Each classification system has its own relative advantages and disadvantages, and no one system is perfect from an imaging standpoint.
OSMOTIC MYELINOLYSIS
Epidemiology
The exact incidence of osmotic myelinolysis is not known. The disorder occurs most commonly in middle-aged, chronically malnourished alcoholic patients, but it is also seen in patients made hyponatremic iatrogenically and in patients with severe liver disease.1
Clinical Presentation
Typically, patients initially present with encephalopathy as a result of hyponatremia. With intravenous sodium replacement, encephalopathy may show clinical improvement, but, subsequently, there is a neurologic decline over the next 48 to 72 hours. Symptoms may include quadriparesis or quadriplegia, pseudobulbar palsy, horizontal gaze palsies, seizures, and coma. Osmotic myelinolysis has an extremely poor prognosis, with only 5% to 10% of patients surviving beyond 6 months. Full recovery from the disorder has been reported, however.2
Pathophysiology
Rapid changes in serum osmolality are believed to cause disruption of the blood-brain barrier, resulting in accumulation of hypertonic fluid in the extracellular space. This ultimately results in extensive noninflammatory demyelination. It is unclear why the pons is preferentially involved, but it has been suggested that the close association of vascular gray matter with orthogonally arranged bundles of myelinated fibers in the pons causes increased susceptibility to compression from cellular edema.2,3
Pathology
Lesions caused by osmotic myelinolysis consist of circumscribed zones of demyelination appearing as regions of discoloration that are abnormally soft and granular, most commonly in the central portions of the upper pons, with involvement of the pontine nuclei and white matter of the base of the pons (Fig. 47-1). Extrapontine sites may also be involved, including the cerebellum, basal ganglia, thalamus, internal and external capsules, and subcortical white matter. The subpial and periventricular regions are spared. Exclusively extrapontine involvement is seen in up to 25% of cases.4
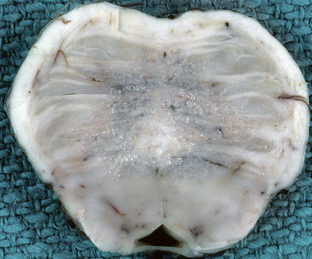
FIGURE 47-1 Central pontine myelinolysis. Axial section through the base of the pons demonstrates discoloration and granularity in the central pons reflecting demyelination. The corticospinal tracts are spared.
(Courtesy of T. W. Bouldin, University of North Carolina, Chapel Hill, NC.)
In the affected areas there is myelin loss with preservation of axons and neuronal cell bodies, which helps in distinguishing these lesions histologically from pontine infarcts. In later stages, infiltration by lipid-laden macrophages may be seen in the central portions of the lesions, with only scant lymphocytes. Oligodendrocytes are lost, but reactive astrocytes may be seen.4
Imaging
CT
CT demonstrates low density in affected areas, reflecting edema. The pons is the most commonly involved site, but lesions may also be seen in the basal ganglia, thalami, subcortical white matter, cerebellum, and middle cerebellar peduncles. The sensitivity of CT for pontine myelinolysis is low, because the pons is frequently obscured by streak artifact from the nearby petrous bone.2
MRI
In the acute phase, MRI demonstrates ovoid areas of T2-weighted (T2W) hyperintensity in the central portion of the pons with sparing of the ventrolateral aspect of the pons and the corticospinal tracts (Fig. 47-2). In some instances, the lesions may appear trident-shaped on axial images. No contrast enhancement is seen after gadolinium administration. In the subacute phase, usually 1 to 2 weeks after onset of symptoms, abnormalities may progress to involve the entire pons. Foci of restricted diffusion may also be evident. In 10% to 50% of cases, extrapontine involvement may be seen in the basal ganglia, thalami, cerebral peduncles, subcortical white matter, cerebellum, and cervicomedullary junction.2,3 MR spectroscopy may demonstrate decreased N-acetyl-aspartate (NAA)/creatine (Cr) and increased choline (Cho)/Cr ratios within the pons. MR perfusion imaging can demonstrate increased perfusion on cerebral blood volume (CBV) maps.5
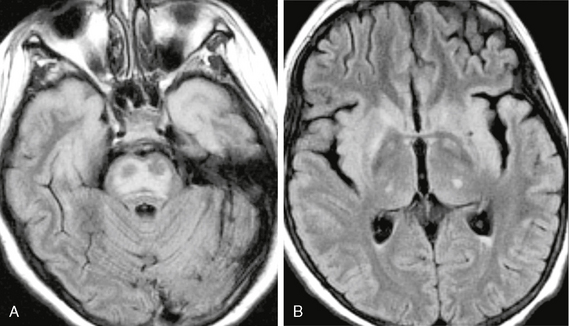
FIGURE 47-2 Osmotic myelinolysis. A, Axial FLAIR image through the pons demonstrates diffuse pontine signal abnormality that spares the periphery of the pons and the corticospinal tracts. The cortex of the anterior temporal lobes are also involved. B, FLAIR image through the basal ganglia shows involvement of the basal ganglia, thalami, claustra, and insula.
Patients who survive demonstrate residual signal abnormality or cavitation in the pons.3
HYPERGLYCEMIC HEMICHOREAHEMIBALLISMUS
Hemichorea-hemiballismus (HCHB) is a syndrome associated with nonketotic hyperglycemia in patients with poorly controlled diabetes mellitus and is characterized by sudden onset of hemiballismus or hemichorea. This syndrome is also called hemiballismus-hemichorea, chorea-ballismus with nonketotic hyperglycemia, and nonketotic hyperglycemia.
Epidemiology
HCHB is seen in poorly controlled diabetic patients with nonketotic hyperglycemia and has been reported mostly in elderly women of Asian origin.6 The entity is quite uncommon, and its exact incidence is not known.
Clinical Presentation
Affected individuals present with acute onset of random, jerking motions in the distal extremities (chorea), or sudden highvelocity flinging or kicking movements (ballismus). Involvement is usually unilateral but may occasionally be bilateral. Most patients will have an established diagnosis of diabetes mellitus, but in some cases HCHB may be the presenting feature of previously undiagnosed diabetes. Blood sugar levels will be elevated and are typically greater than 200 mg/dL. Serum osmolarity may be normal or mildly elevated.7 Symptoms are usually reversible with treatment of hyperglycemia, but the condition may rarely progress to coma and death. Chorea and ballismus are nonspecific symptoms and can also result from other conditions, including cerebrovascular disease, infections, drug toxicity, other metabolic disorders, neurodegenerative diseases, autoimmune disorders, and tumors.
Pathophysiology
Hyperglycemia results in global decreased regional cerebral blood flow, which is maximal in the basal ganglia. It is presumed that hyperglycemia and hyperviscosity of blood in diabetic patients causes ischemia in the striatum.6 The decreased flow may contribute to decreased local amounts of the neurotransmitter γ-aminobutyric acid (GABA). Decreases in striatal GABA may allow increased pallidal activity, with resultant dyskinesia affecting the contralateral side of the body.7
The explanation for the specific imaging changes seen in HCHB remains controversial. Some believe that hyperglycemia causes partial ischemia in the basal ganglia with resultant microhemorrhage or reversible calcium influx. Alternative explanations include manganese accumulation in reactive astrocytes after ischemia, which would explain hyperintensity on T1-weighted (T1W) MR images but not high density on CT. Still others suggest protein desiccation during the course of wallerian degeneration is responsible for the imaging abnormalities.8
Histologically, the abnormal putamen contains multiple infarcts with reactive astrocytosis, fragmentation of axons, and myelin pallor. Punctate calcifications and scattered microhemorrhages may be seen but are an inconstant finding.6
Imaging
CT
CT in patients with HCHB classically demonstrates increased density in the caudate, putamen, or both. Involvement is typically unilateral but can be bilateral. When symptoms are unilateral, the lesions are located in the contralateral corpus striatum. Lesions typically resolve over time after treatment.7
MRI
MRI will typically demonstrate increased signal intensity in one of the basal ganglia on T1W images (Fig. 47-3).7 Bilateral basal ganglia involvement can occasionally be seen. Hyperintensity on diffusion-weighted images with corresponding reduced apparent diffusion coefficients (ADC) may also be seen but may be reversible. Other entities that may produce hyperintensity in the basal ganglia on T1W imaging are manganese poisoning, prolonged parenteral nutrition (presumedly due to deposition of manganese), and chronic liver failure.
DISORDERS OF IRON AND COPPER METABOLISM
Neurodegeneration with Brain Iron Accumulation
Epidemiology
The exact prevalence of NBIA is not known. The disease occurs with equal frequency in males and females, and no race predilection has been identified. Although NBIA is typically a disease of childhood, symptom onset during adulthood has been reported. The classic form of the disease usually presents by the age of 10. The atypical form presents in the second decade of life and occasionally in early adulthood.9
Clinical Presentation
The early-onset form of the disease presents in childhood with dystonia, dysarthria, rigidity, and choreoathetosis. Symptoms related to involvement of the corticospinal tract (spasticity, hyperreflexia) are also frequently observed. The most common presenting symptoms are gait or postural difficulties. In addition, most patients with the classic form of disease have clinical or electroretinographic evidence of retinopathy due to pigmentary degeneration of the retina. Symptoms progress rapidly to severe disability by the age of 20, ultimately culminating in early death.
In the atypical form of the disease, symptoms are much more variable. Extrapyramidal symptoms occur less frequently than in the classic form, and the degree of dystonia and rigidity is generally less severe and progresses more slowly. Patients may remain ambulatory for up to 40 years after onset of symptoms. Spasticity, hyperreflexia, and clinical evidence of retinopathy are also less common. In a significant number of patients with the atypical form of disease, difficulty with speech may be the sole presenting feature, unlike patients with the classic form who almost never present initially with dysarthria (although they may develop it later in the disease course). Cognitive decline eventually resulting in dementia is prominent in atypical NBIA but is rare in the classic form.9
Pathophysiology
In the majority of patients with NBIA, the disease has been linked to a mutation in the PANK2 gene located on chromosome 20p13. PANK2 encodes a pantothenate kinase involved in the biosynthesis of coenzyme A from vitamin B5. PANK2 mutations are shown in all cases of classic NBIA and approximately one third of cases of the atypical form.9 Although the genetic mutation causing most cases of the disorder has been identified, the exact mechanism of tissue injury is not known. It has been hypothesized that deficiency of PANK2 leads to accumulation of cysteine-containing neurotoxic compounds in highly sensitive regions of the brain, resulting in tissue damage and edema. Accumulation of excess iron in normally iron-rich brain structures is suspected to be secondary to tissue damage in the disease.10
Pathology
Macroscopically, rusty-brown discoloration of the globus pallidus (particularly the internal segment) (Fig. 47-4) and reticular zone of the substantia nigra is seen.4
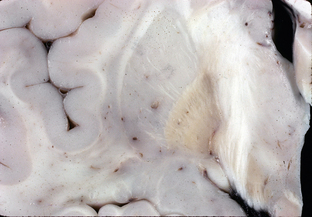
FIGURE 47-4 Neurodegeneration with brain iron accumulation. Coronal section through the basal ganglia demonstrates rust-brown discoloration of the globus pallidus, most pronounced in its internal segment.
(Courtesy of T. W. Bouldin, University of North Carolina, Chapel Hill, NC.)
On microscopy, brownish yellow pigment is seen in the cytoplasm of nerve cells, microglia, and astrocytes from the globus pallidus (internal segment) and substantia nigra. Extracellular pigment is also present around blood vessels in the same areas. Pigment deposits give a strong reaction for iron and are periodic acid–Schiff (PAS) positive and Sudan black positive. Neuronal depletion is seen in the globus pallidus, in the reticular zone of the substantia nigra, and occasionally in the subthalamic nuclei and cerebellum. Reactive gliosis is also seen. A characteristic finding is the presence of rounded or oval structures known as spheroids, which represent axonal swellings.4
Imaging
CT
CT is generally not helpful in the diagnosis of NBIA. Although frequently normal, may demonstrate cerebral or cerebellar atrophy and mineralization of the basal ganglia, particularly the globus pallidus, probably reflecting deposition of iron-staining pigment.11
MRI
Findings on MRI in patients with NBIA correlate with the presence or absence of PANK2 mutations. In patients with the PANK2 mutation, the characteristic finding on MRI is the “eye of the tiger” sign, which consists of T2 signal hypointensity in the globus pallidus with a central region of T2 hyperintensity (Fig. 47-5). The low signal intensity of the globus pallidus reflects excess iron deposition, and the focus of hyperintensity centrally represents areas of loose tissue with vacuolization.12 Enhancement in the globus pallidus after contrast agent administration is not seen. The earliest imaging finding is isolated T2 hyperintensity in the globus pallidus interna. Patients with this finding typically go on to develop the “eye of the tiger” sign within 3 years. Hypointensity in the reticular layer of substantia nigra on T2W images may also be seen in these patients.10
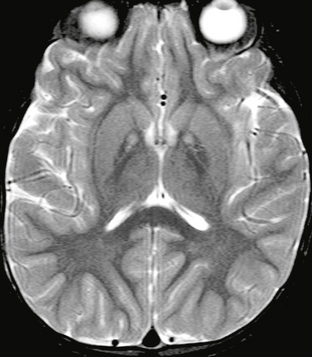
FIGURE 47-5 Neurodegeneration with brain iron accumulation. Axial T2W MR image demonstrates symmetric foci of hyperintensity on a background of hypointensity of the globi pallidi. This is an example of the “eye of the tiger” sign.
Patients without the PANK2 mutation will not demonstrate the “eye of the tiger” sign. These patients may show T2 hypointensity alone in the globus pallidus. Cerebral and cerebellar atrophy and iron deposition in the red nucleus and dentate nucleus are common features in patients without the PANK2 mutation.9
Wilson’s Disease
Wilson’s disease is a rare, autosomal recessive defect of copper metabolism that causes accumulation of abnormal amounts of copper in various tissues, with a predilection for involvement of the brain, kidney, and liver. Manifestations of Wilson’s disease include liver disease and neurologic symptoms. Alternate names include hepatolenticular degeneration, progressive lenticular degeneration, and Westphal-Strümpell pseudosclerosis.
Clinical Presentation
Diagnosis of Wilson’s disease is usually confirmed on the basis of Kayser-Fleischer rings, decreased serum copper and ceruloplasmin levels, and increased 24-hour urinary copper excretion. Patients require lifelong treatment with de-coppering agents such as penicillamine, trientine, and zinc.14,15
Pathophysiology
The genetic defect responsible for Wilson’s disease has been linked to chromosome 13q14.3. The gene ATP7B encodes a transmembrane protein, which plays a role in the transport of copper into cells, incorporation of copper into the plasma protein ceruloplasmin, and excretion of excess copper stores in the bile. Defects in ATP7B result in excess copper accumulation in tissues, most notably the liver and the brain.15 Excess copper may combine with sulfhydryl, carboxyl, or amine groups and cause improper enzymatic activity, ultimately damaging cell structure. In addition, increased extracellular copper can cause oxidative stress, which leads to cell death.16
Pathology
On autopsy, the brain may appear normal externally, but on sectioning there will be shrinkage and brownish discoloration of the putamen (particularly the middle third) and caudate nuclei. Cysts may be present in the putamen, and the cerebral white matter may be softened and cystic. The globus pallidus, subthalamic nucleus, thalamus, and brain stem are often involved but less severely. Spongy degeneration in the cerebral cortex and white matter may also be seen.4
On histology, the brain will demonstrate an increase in the number of astrocytes (known as Alzheimer type 2 cells) within gray matter, swollen glia, liquefaction, and areas of spongiform degeneration, which is most pronounced in the putamina. Alzheimer type 2 cells are notable for enlarged, vesicular nuclei, and prominent nucleoli with inconspicuous cytoplasm. Macrophages may be seen within cystic regions and in nearby perivascular spaces. Perivascular and parenchymal granular copper deposits are evident on rhodanine or rubeanic acid stains.17 Neuronal loss and gliosis may be seen. Characteristic of Wilson’s disease is the presence of Opalski cells—large cells containing fine granular cytoplasm and small, slightly abnormal nuclei—that are believed to represent degenerating astrocytes.15,17
Imaging
CT
On CT, patients with Wilson’s disease demonstrate generalized atrophy. Hypodense lesions may be evident in the basal ganglia and frontal white matter. In general, CT is less sensitive than MRI for detecting gray and white matter abnormalities.18
MRI
Characteristic MRI findings in Wilson’s disease are T1 hypointense and T2 hyperintense lesions, most commonly in the basal ganglia, with involvement most frequently in the putamina, followed by the caudate and globus pallidus. Lateral putaminal involvement is a characteristic feature. Thalamic involvement is also common and typically affects the lateral nuclei with relative sparing of the dorsomedial nuclei. Cerebellar involvement may also be seen, particularly in the superior and middle cerebellar peduncles. Involvement of the brain stem, in particular the midbrain, is also common and may be limited to the dorsal or periaqueductal regions (Fig. 47-6). Contrast enhancement with gadolinium is not typical.18
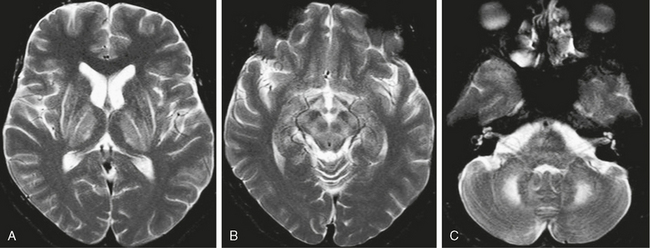
FIGURE 47-6 Wilson’s disease. A, Axial T2W MR image through the basal ganglia demonstrates symmetric high signal intensity within the lentiform nuclei and thalamus. Hyperintensity is particularly marked along the lateral margins of the putamina. Note that the dorsomedial portions of the thalami are spared. B, Axial T2W MR image through the level of the midbrain demonstrates high signal intensity in the midbrain with superimposed low signal intensity within the red nuclei and substantia nigra, giving the characteristic “panda sign.” C, Axial image through the level of the middle cerebellar peduncles demonstrates bilateral cerebellar and middle cerebellar peduncle hyperintensities.
One sign considered characteristic of Wilson’s disease is the “panda sign” in which T2W images demonstrate hyperintensity in the midbrain superimposed on low signal in the substantia nigra and red nucleus, giving the appearance of a panda face (see Fig. 47-6B). Pontine involvement with features similar to central pontine myelinolysis can also be seen.14 Atrophy is common; and in addition to deep gray matter involvement, abnormalities in the cerebral white matter may be seen in approximately 25% of patients with Wilson’s disease. White matter lesions are usually T2 hyperintense, are asymmetric, and have a frontal lobe predilection.14 Signal changes have been shown to reverse with treatment, and improvement on follow-up MRI has been shown to correlate with clinical response to treatment.16
One report of MR spectroscopy in a patient with Wilson’s disease describes the presence of lactate, a decreased NAA/Cr ratio, and a markedly increased ADC in a putaminal lesion.14
Patients presenting primarily with hepatic disease may demonstrate T1W hyperintensities in the striatal regions, as can be seen in other forms of chronic liver failure.14
DISORDERS RELATED TO ETHANOL ABUSE
Ethanol is the most widely used substance of abuse worldwide. In the United States, the prevalence of alcohol abuse is approximately 6% among males and 2% among females.3 Long-term alcohol abuse is known to induce selective neuronal damage, but the exact mechanism of injury (direct toxicity vs. toxicity of breakdown products) is unknown. Among the most commonly recognized effects of chronic alcohol abuse seen on neuroimaging are cerebral atrophy and cerebellar atrophy. Cerebellar atrophy has a greater correlation with alcohol use and is typically more marked in the rostral vermis and adjacent superior cerebellar surfaces (Fig. 47-7). In addition, demyelinating lesions in the cerebral white matter, similar to those seen in multiple sclerosis, have been described. The pathogenesis of demyelination in these cases is unknown.2
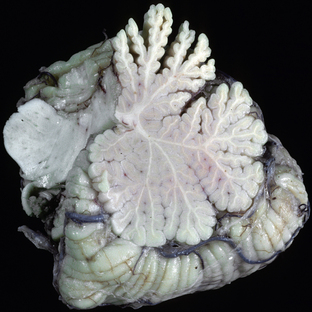
FIGURE 47-7 Cerebellar atrophy associated with chronic ethanol abuse. Sagittal section through the cerebellar vermis demonstrates atrophy predominantly involving the rostral vermis.
(Courtesy of T. W. Bouldin, University of North Carolina, Chapel Hill, NC.)
A common finding in patients with alcoholic cirrhosis and other forms of liver failure is symmetric high-signal intensity in the basal ganglia (Fig. 47-8). This finding can be seen in the absence of signs of hepatic encephalopathy and is believed to be caused by the deposition of paramagnetic substances, including copper and manganese, which have bypassed the detoxification system of the liver. Manganese poisoning, which typically presents as levodopa-resistant parkinsonism, and prolonged total parenteral nutrition (which is known to increase serum levels of manganese) also demonstrate similar basal ganglia hyperintensities on T1W MRI.19
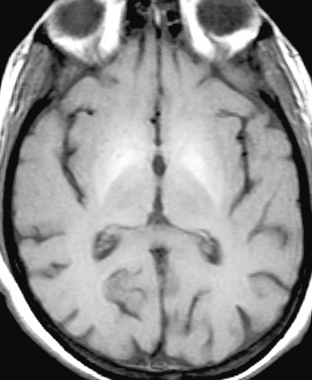
FIGURE 47-8 Liver failure. Axial unenhanced T1W image demonstrates symmetric hyperintensity in the lentiform nuclei, most pronounced in the globi pallidi.
Wernicke Encephalopathy
Wernicke encephalopathy is a neurologic disorder that results from chronic thiamine deficiency. The disease is characterized by ocular abnormalities, ataxia, and confusion.3 Korsakoff psychosis, also caused by chronic thiamine deficiency, is frequently seen in conjunction with Wernicke encephalopathy and is characterized by more severe cognitive dysfunction and memory loss. The combination of these two entities is known as Wernicke-Korsakoff syndrome.
Epidemiology
The prevalence of Wernicke encephalopathy–specific changes at autopsy may be as high as 2.8% in the general population.3,20 It is estimated that up to 80% of patients with the syndrome go undiagnosed.20 Wernicke encephalopathy is predominantly a disease of alcoholics, but any condition that results in thiamine deficiency, including anorexia nervosa, gastrointestinal disorders, protracted parenteral therapy, hyperemesis gravidarum, AIDS, and hematologic malignancies, may cause it. Males are more likely to be affected, owing to the higher prevalence of alcoholism in the male population.
Clinical Presentation
Onset of symptoms may be abrupt or gradual. A classic triad of ocular abnormalities, ataxia, and confusion is seen in approximately 30% of cases.3 Ocular disturbances may include nystagmus, abducens and conjugate gaze palsies, and ophthalmoplegia. Symptom improvement occurs with thiamine replacement.
If treatment with thiamine is not promptly initiated, the more severe symptoms of Korsakoff psychosis occur. The characteristic feature of Korsakoff psychosis is antegrade and retrograde amnesia, leading to confabulation. Some consider Korsakoff psychosis to be a chronic variant of Wernicke encephalopathy. If not treated, Wernicke encephalopathy has a mortality of up to 20%.3
Pathophysiology
Thiamine is involved in a number of cellular processes, including intermediate carbohydrate metabolism, maintenance of membrane integrity and osmotic gradients across cell membranes, redox equilibrium, and nucleic acid synthesis. Depletion of thiamine leads to cerebral lactic acidosis and edema with swelling of astrocytes, oligodendrocytes, myelin fibers, and dendrites. The periventricular regions utilize thiamine-dependent glucose and oxidative metabolism and are therefore susceptible to injury when thiamine deficiency occurs.3
Pathology
Lesions that are visible macroscopically as areas of congestion and petechial hemorrhage are seen in the subependymal regions around the third ventricle and aqueduct, the floor of the fourth ventricle, the pulvinar and dorsomedial thalamic nuclei, hypothalami, superior cerebellar vermis, and mammillary bodies (Fig. 47-9). In the later stages these areas will demonstrate brown discoloration and softening.3,17
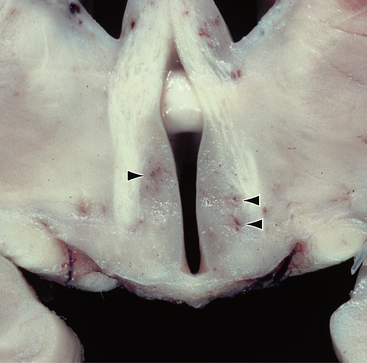
FIGURE 47-9 Wernicke encephalopathy. Coronal section through the third ventricle demonstrates petechial hemorrhages along the walls of the third ventricle within the hypothalami (arrowheads).
(Courtesy of T. W. Bouldin, University of North Carolina, Chapel Hill, NC.)
In the acute stages, focal capillary dilatation, congestion, and endothelial swelling will be seen microscopically. Petechial hemorrhages and perivascular macrophages are also commonly seen. In the subacute and later stages, histologic findings include demyelination, fibrillary gliosis, capillary endothelial hyperplasia and proliferation, and petechial hemorrhages. Remyelination can occur if thiamine deficiency is corrected in the early stage. Neurons are relatively spared but may be shrunken. In the thalamus (most commonly the dorsomedial thalamic nuclei) there may be neuronal shrinkage without capillary changes.3,17
Imaging
CT
CT is generally not helpful in the diagnosis of acute Wernicke encephalopathy. Rarely, low density may be seen in the periventricular regions of the thalamus.21
MRI
MRI in acute Wernicke encephalopathy will demonstrate T2W hyperintensities located in the walls of the third ventricles, the pulvinars and dorsomedial nuclei of the thalami, the periaqueductal gray matter, the pineal regions, the floor of the fourth ventricle, and the mammillary bodies. Enhancement of these regions may be seen, and enhancement of the mammillary bodies is considered pathognomonic for Wernicke encephalopathy (Fig. 47-10). Microhemorrhages may be evident. Diffusion-weighted images may demonstrate hyperintensity with decreased ADCs in the involved regions, reflecting cytotoxic edema.3
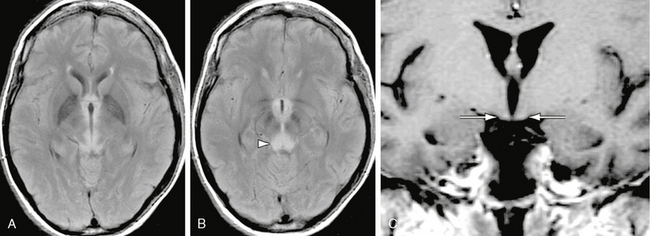
FIGURE 47-10 Wernicke encephalopathy. A, Axial FLAIR image demonstrates increased signal in the medial thalami. B, Axial FLAIR image inferior to A demonstrates increased signal in the periaqueductal gray matter (arrowhead). C, Coronal gadolinium-enhanced T1W image demonstrates faint enhancement of the mammillary bodies (arrows).
In patients with chronic Wernicke encephalopathy, signal abnormalities in the aforementioned regions may not be evident but there will be diffuse brain atrophy, particularly involving the fornices and mammillary bodies, with enlargement of the third ventricle.3
Marchiafava-Bignami Disease
Epidemiology
Marchiafava-Bignami disease is a very rare condition and occurs primarily in middle-aged chronic alcoholics. The mean age at diagnosis is in the mid 40s. Males are more commonly diagnosed with the disease than females,22 which is probably attributable to the higher rate of alcoholism among males. Rare cases have been reported in nonalcoholic patients.
Clinical Presentation
The disease may present in two forms. The acute form presents as marked neurologic changes, including seizures and coma, and is often fatal. In the chronic form of the disease, patients present with nonspecific neurologic signs, including cognitive impairment, gait disturbances, limb hypertonia, and dysarthria. The characteristic findings are signs of interhemispheric disconnection.2,3 In some cases, Marchiafava-Bignami disease may be difficult to distinguish from Wernicke encephalopathy and the two entities may coexist in the same patient.
Pathophysiology
The exact pathogenesis of tissue damage in Marchiafava-Bignami disease is not known. It is believed that a toxic agent contained in inexpensively produced red wines and malnutrition (particularly deficiencies of certain B vitamins) may play a role in the development of the disease.3
Pathology
Grossly, there may be edema, necrosis, and occasional hemorrhage in the corpus callosum. A pattern of necrosis is seen in which the middle layer of the corpus callosum degenerates (layered necrosis), which is a typical finding in the disease.3 In the chronic phase, there is thinning of the corpus callosum, which may be focal.2,3
A well-demarcated band of demyelination is present in the lesions of the corpus callosum, and lesions may extend to involve the neighboring white matter of the centrum semiovale. Other white matter tracts including the anterior commissure and middle cerebellar peduncles may also be affected.17
Demyelination is seen, with relative, but incomplete, axonal sparing. There is reactive gliosis and vascular proliferation in the affected areas.17