LEARNING OBJECTIVES
1. List an appropriate differential diagnosis for upper lung disease seen on chest radiography or computed tomography (CT).
2. Describe the radiographic classification of sarcoidosis.
3. State the three most common locations (Garland triad) for adenopathy to occur in the chest of patients with sarcoidosis.
4. List four common etiologies of “eggshell” calcified lymph nodes in the chest.
5. Recognize progressive massive fibrosis secondary to silicosis on chest radiography and CT.
6. Recognize and describe the typical appearance of cystic fibrosis on chest radiography and CT.
7. Describe the radiologic manifestations of primary pulmonary tuberculosis.
8. Name the most common segmental sites of involvement for reactivation tuberculosis in the lung.
9. Define a Ghon lesion (calcified pulmonary parenchymal granuloma) and Ranke complex (calcified node and Ghon lesion); recognize both on a chest radiograph and CT and describe their significance.
10. Suggest the possibility of radiation as a cause of new upper lung opacification on a chest radiograph of a patient with evidence of mastectomy and/or axillary node dissection or known head and neck cancer.
11. Describe the acute and chronic phases of radiation-caused changes in the lungs, including the time course and typical chest radiograph and CT appearances.
12. Recognize the typical appearance of irregular lung cysts on chest CT of a patient with Langerhans cell histiocytosis.
13. Name the major categories of disease that cause chest radiographic or CT abnormalities in the immunocompromised patient.
14. Other than typical bacterial infection, name two important infections and two important neoplasms to consider in patients with acquired immunodeficiency syndrome (AIDS) and chest radiographic or CT abnormalities.
15. Describe the typical chest radiographic and CT appearances of Kaposi sarcoma.
16. Describe the chest radiographic and CT appearances of Pneumocystis jiroveci pneumonia.
17. Name four important etiologies of hilar and mediastinal lymphadenopathy in patients with AIDS.
18. Describe the time course and chest radiographic appearance of a blood transfusion reaction.
19. Describe the chest radiographic and CT appearances of a miliary pattern and provide a differential diagnosis.
20. Name and describe the types of pulmonary Aspergillus disease.
21. Identify an intracavitary fungus ball on chest radiography and CT.
22. Name the most common pulmonary infections that occur after solid organ (e.g., liver, renal, lung, cardiac) and bone marrow transplantation.
23. Describe the chest radiographic and CT findings of posttransplant lymphoproliferative disorders.
Pulmonary infections are a major cause of morbidity and mortality, especially in immunocompromised patients. Immunocompromised patients have altered immune mechanisms and are predisposed to opportunistic infections. Numerous factors are associated with an immunocompromised state, including but not limited to diabetes; renal or liver failure; advanced age; bone marrow or solid organ transplantation; acquired immunodeficiency syndrome (AIDS); presence of access lines (e.g., intravenous lines, endotracheal tubes, chest tubes); splenectomy, hospital environment (predisposing to nosocomial pneumonia); underlying malignancy; drug therapy (e.g., steroids, chemotherapy); and immune deficiencies (e.g., hypogammaglobulinemia). Some of the clinically important infections and other diseases seen in immunocompetent and immunocompromised patients tend to have an upper lung–predominant distribution (e.g., mycobacterial and fungal disease). Recognition of an upper lung distribution of disease helps the clinician to form an appropriate differential diagnosis. This chapter begins with a discussion of upper lung disease, including infectious and noninfectious causes, and continues with a review of the disorders that occur in immunocompromised individuals and their radiographic appearances.
UPPER LUNG DISEASE
Upper lung refers to the upper one-third of the lung, which includes the majority of the upper lobes and the uppermost portion of the superior segments of the lower lobes. In the normal upright lung, blood flow and ventilation predominate in the lung base; in many lung disorders however, the greatest degree of abnormality occurs in the upper lung. Alterations in ventilation–perfusion, lymphatic flow, metabolism, and mechanics are proposed as pathogenic factors in upper lung localization of lung disease (1). Two mnemonics, “SHRIMP” and “CASSET,” can be used to recall common and uncommon disorders occurring in the upper lungs (Table 10.1). Because it may be difficult to appreciate a predominantly upper lung distribution of disease on chest radiography, it is useful to consider the differential diagnoses given in Table 10.1, even if the disease appears diffuse, any time the upper lungs are as affected as much or more than the middle and lower lungs.
SARCOIDOSIS
Sarcoidosis is a common systemic disease of unknown etiology characterized by widespread development of noncaseating granulomas. These granulomas are nonspecific and resemble those in many other granulomatous processes, except for tuberculosis (TB), a disease in which caseous necrosis of granulomas is usually seen. Sarcoidosis is 10 times more common in African-Americans than in Caucasians (2). Most patients who present with sarcoidosis are between the ages of 20 and 40, but the disease occurs as early as 1 year and as late as 80 years of age (3). The disease is 2 to 3 times more common in African-American women than in African-American men (3). The lung is the most commonly involved organ in patients with sarcoidosis and accounts for most of the morbidity and mortality, with an overall mortality rate between 2.2% and 7.6% (3).
Table 10.1 UPPER LUNG DISEASE
“SHRIMP”
Sarcoidosis
Histiocytosis, Langerhans cell
Radiation pneumonitis (cancers of head/neck and breast)
Infection (tuberculous, fungal)
Metastasesa
Pneumoconiosesb (silicosis, coal miner’s)
“CASSET”
Cystic fibrosis
Ankylosing spondylitis
Silicosis
Sarcoidosis
Eosinophilic granulomatosis (Langerhans cell histiocytosis)
Tuberculous, fungal infection
Table 10.2 CLASSIFICATION OF SARCOIDOSIS ON CHEST RADIOGRAPHY
0 | Normal chest radiograph |
I | Hilar or mediastinal nodal enlargement only |
II | Nodal enlargement and parenchymal disease |
III | Parenchymal disease only |
IV | End-stage lung (pulmonary fibrosis) |
Sarcoidosis can be classified according to its appearance on the chest radiograph (Table 10.2) (4). Patients commonly, but not necessarily, progress through each class, and the class at presentation can, but does not always, correlate with prognosis (5). Forty-five percent to 65% of patients are class I at the time of presentation. Lymphadenopathy is the most common intrathoracic manifestation of sarcoidosis and occurs in 75% to 80% of patients at some point in their illness (6). The classic pattern of lymphadenopathy is bilateral hilar and right paratracheal nodal enlargement, the so-called Garland triad or 1-2-3 sign (Figs. 10.1 and 10.2), although any mediastinal nodes can be and frequently are involved. The hilar lymph nodes are usually symmetric in appearance and can be massively enlarged (“potato nodes”) but are usually clear of the cardiac borders, a feature that helps distinguish sarcoidosis from lymphomatous lymphadenopathy, as the latter usually abuts the cardiac margins. Of patients with class I disease at initial examination, about 60% go on to complete resolution (7), with parenchymal disease occurring in the remaining patients. Nodal calcification is seen in up to 20% of cases (8), and in some of these cases (approximately 5%), the calcification is of a peripheral “eggshell” pattern (Figs. 10.3 and 10.4). Eggshell calcification is largely limited to sarcoidosis and silicosis, but it can be seen in other disorders (Table 10.3) (9).
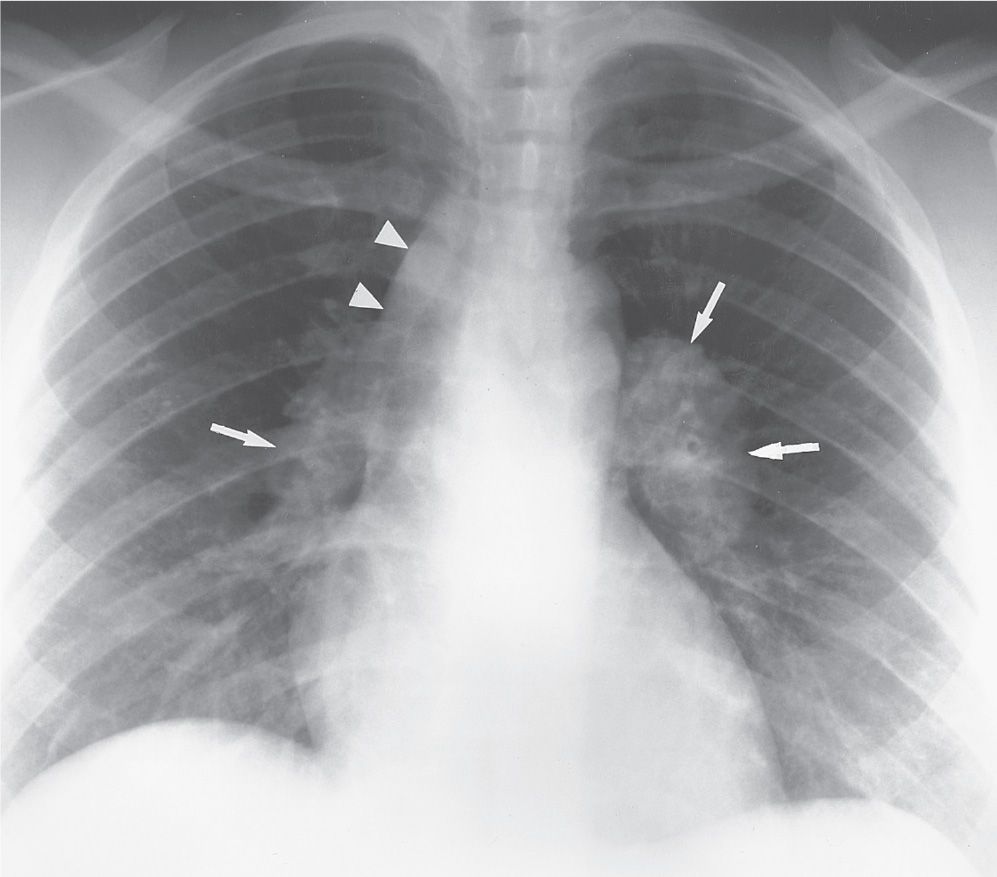
FIG. 10.1 • Sarcoidosis. Posteroanterior (PA) chest radiograph of a 31-year-old woman with class I sarcoidosis shows right paratracheal (arrowheads) and bilateral hilar (arrows) lymphadenopathy. This pattern of lymphadenopathy is classic for sarcoidosis and is referred to as the 1-2-3 sign or Garland triad.
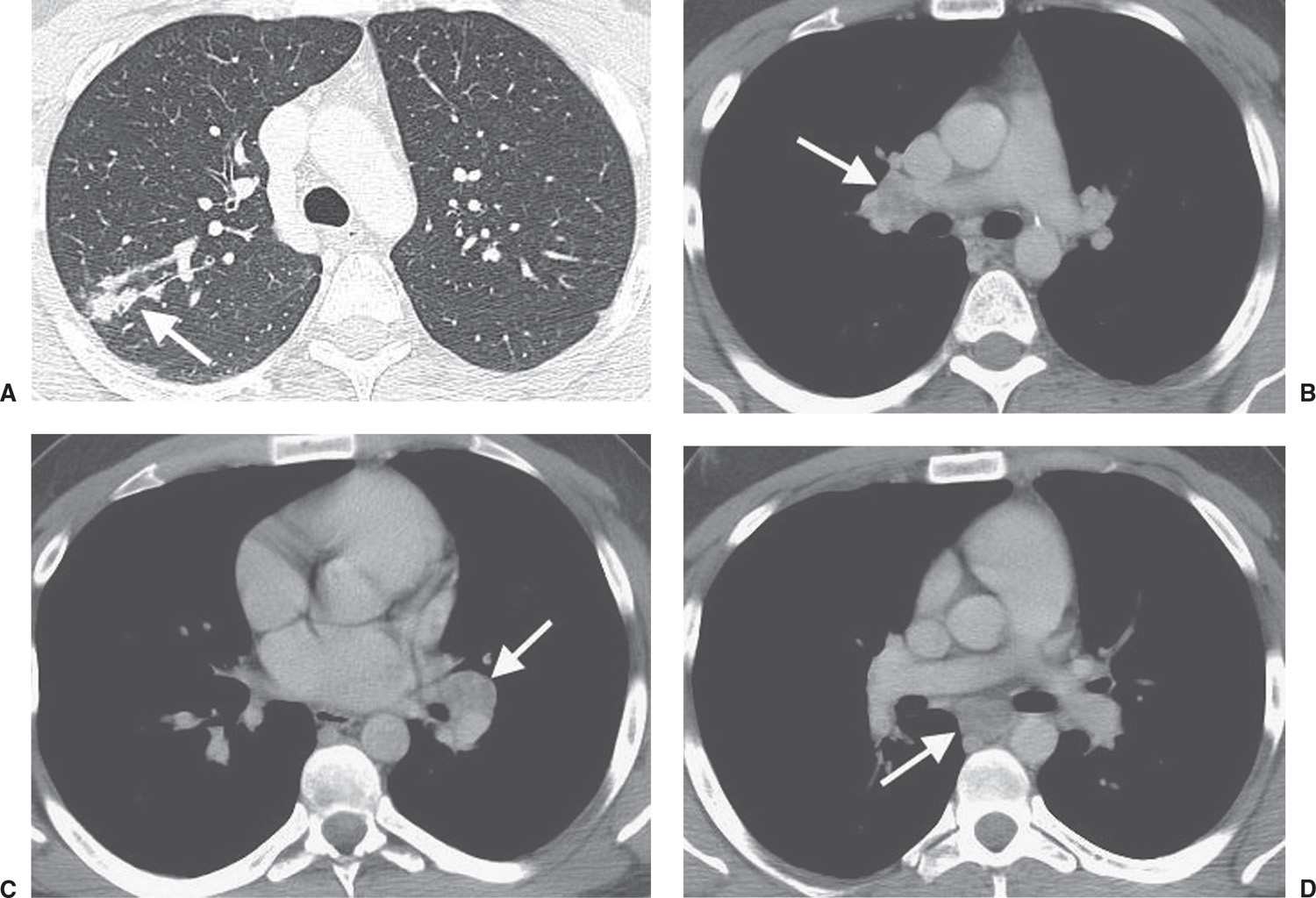
FIG. 10.2 • Sarcoidosis. A: CT of a 23-year-old woman shows ill-defined nodules in a bronchovascular distribution (arrow) in the right upper lobe. B: CT with mediastinal windowing shows right hilar lymphadenopathy (arrow). C: CT at the level of the inferior pulmonary veins shows left hilar lymphadenopathy (arrow). D: CT at the level of the lower lobe pulmonary arteries shows subcarinal lymphadenopathy (arrow).
Parenchymal disease is seen on chest radiography at the time of presentation in approximately half of patients with sarcoidosis. Radiographic patterns of parenchymal disease include reticulonodular opacities, ill-defined opacities that have an appearance of alveolar filling, large nodules, and lung fibrosis. Reticulonodular opacities are the most common pattern, seen in 75% to 90% of patients with parenchymal disease; the opacities are usually bilaterally symmetric with a distribution predominantly in the middle and upper lungs (10) (Figs. 10.5 and 10.6). In 10% to 20% of patients, opacities with airspace features develop, which can be ill-defined or focal, nodular, and well-defined. The term alveolar sarcoid refers to this pattern, although the “airspace” disease represents an interstitial process that compresses and obliterates alveoli. Alveolar sarcoid generally consists of bilateral, multifocal, poorly defined opacities showing a predilection for the peripheral lungs (11) (Fig. 10.7). The peripheral distribution is particularly well seen with computed tomography (CT).
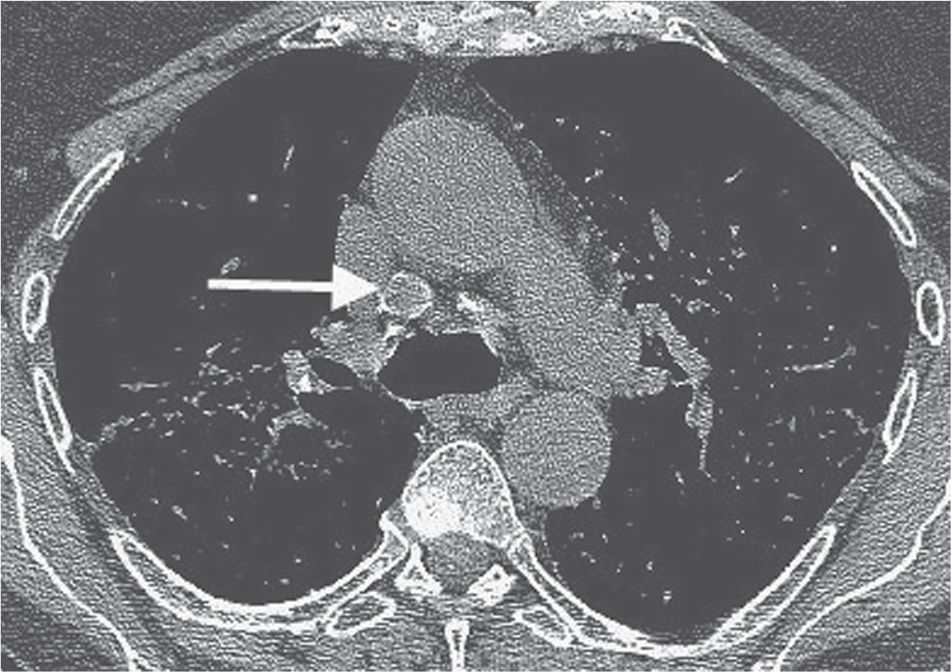
FIG. 10.3 • Sarcoidosis. CT shows precarinal lymphadenopathy with rim calcification (arrow). This pattern of calcification is referred to as “eggshell” calcification and is seen in approximately 5% of patients with sarcoidosis.
Sarcoid granulomas may resolve completely or heal by fibrosis. Pulmonary fibrosis occurs in approximately 20% of patients with sarcoidosis, and the radiologic features are considered by some authors to be almost pathognomonic. The findings consist of permanent, coarse, linear opacities radiating laterally from the hilum into adjacent upper and middle lungs. Bullae can form in the upper lungs. The hila are pulled upward and outward, and vessels and fissures are distorted. The fibrosis is occasionally so severe that massive parahilar opacities in the middle and upper lungs, resembling those of progressive massive fibrosis of silicosis, are seen (Fig. 10.8).
CT can define the anatomic location of parenchymal sarcoid granulomas much more accurately (12–14). The most common finding of sarcoidosis on CT is multiple, 1- to 5-mm nodules, usually with irregular margins, in a perilymphatic distribution (bronchovascular margins, along interlobular septa, subpleurally, and in the center of secondary pulmonary lobules) (Fig. 10.9). Septal thickening from sarcoidosis has a beaded appearance, a feature that helps distinguish it from pulmonary edema, in which the septal thickening is typically smooth. Patchy ground-glass opacities are seen in about 50% of patients with sarcoidosis and rarely may be the only CT abnormality (Fig. 10.10). Fibrosis is better characterized on CT than on chest radiography. CT can show findings of sarcoidosis when the chest radiograph is normal, and patients can have a normal CT study yet have sarcoidosis proved by lung biopsy (13).
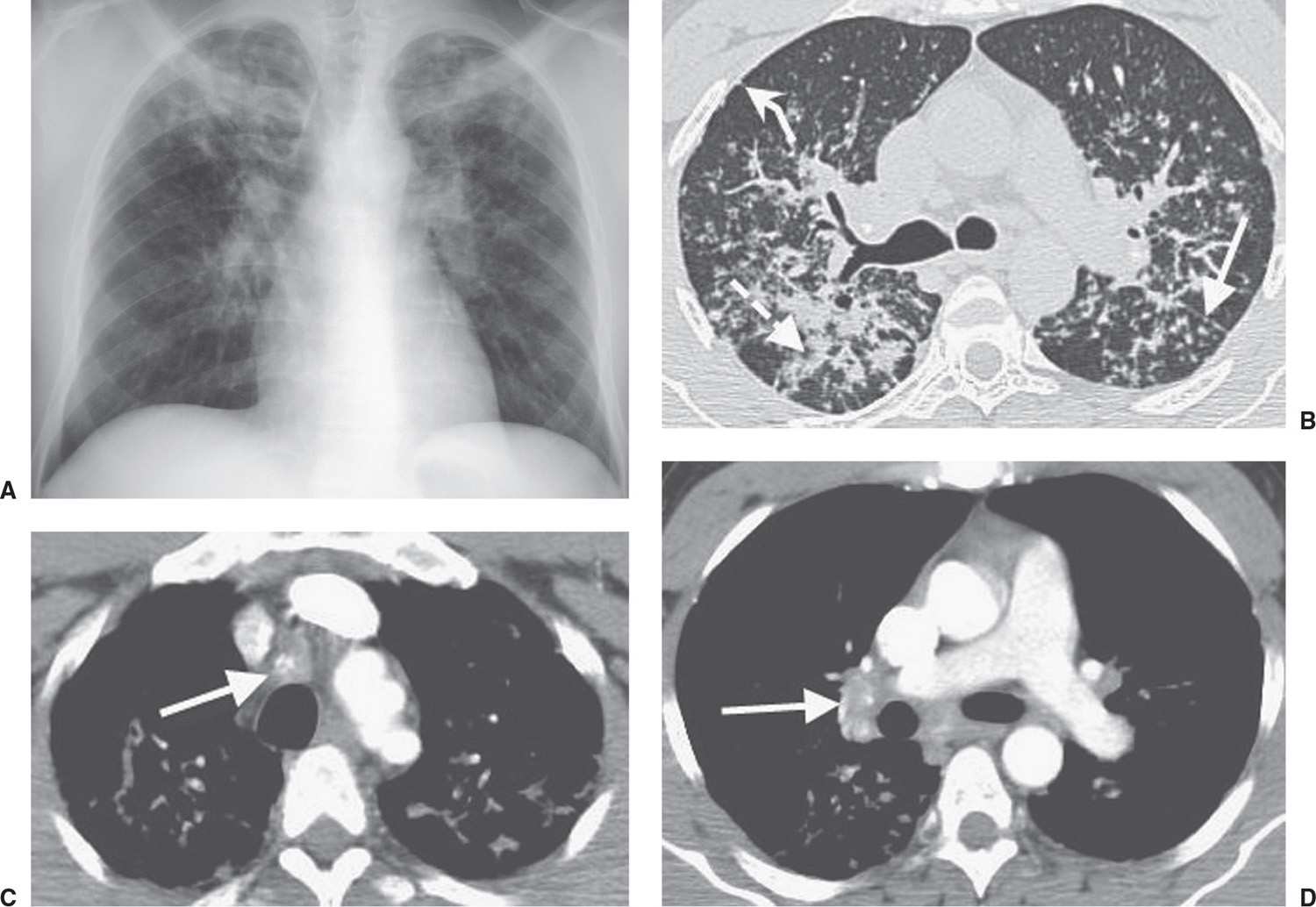
FIG. 10.4 • Sarcoidosis. A: PA chest radiograph of a 37-year-old man shows bilateral upper lobe nodular disease and hilar enlargement (class II). B: CT shows nodules of varying size along the fissures (straight solid arrow) and bronchovascular bundles (dashed arrow) and in a subpleural location (curved solid arrow). C: CT with mediastinal windowing shows central calcification of right paratracheal lymph nodes (arrow). D: CT at a lower level shows calcification of right hilar nodes (arrow).
Table 10.3 COMMON CAUSES OF “EGGSHELL” CALCIFICATION OF LYMPH NODES IN THE CHEST
“SIT”
Sarcoidosis
Silicosis
Infection (tuberculous, fungal)
Treated lymphoma
Fungus balls (mycetomas) can develop in cystic areas that develop from sarcoidosis, and sarcoidosis is the second most common predisposing condition (afterTB) leading to the development of mycetoma (15). Hemoptysis resulting from mycetoma formation can be life-threatening. Mycetomas occur in the upper lobes and should be suspected when new opacities are seen in an area of chronic cystic or bullous disease, especially when they are accompanied by new apical pleural or extrapleural opacity on chest radiography. There are myriad other atypical features of sarcoidosis, including pleural effusions, pleural thickening, cavitary nodules, bronchostenosis, pulmonary artery hypertension from periarterial granulomatosis (Fig. 10.11), cor pulmonale, and pneumothorax from chronic fibrosis.
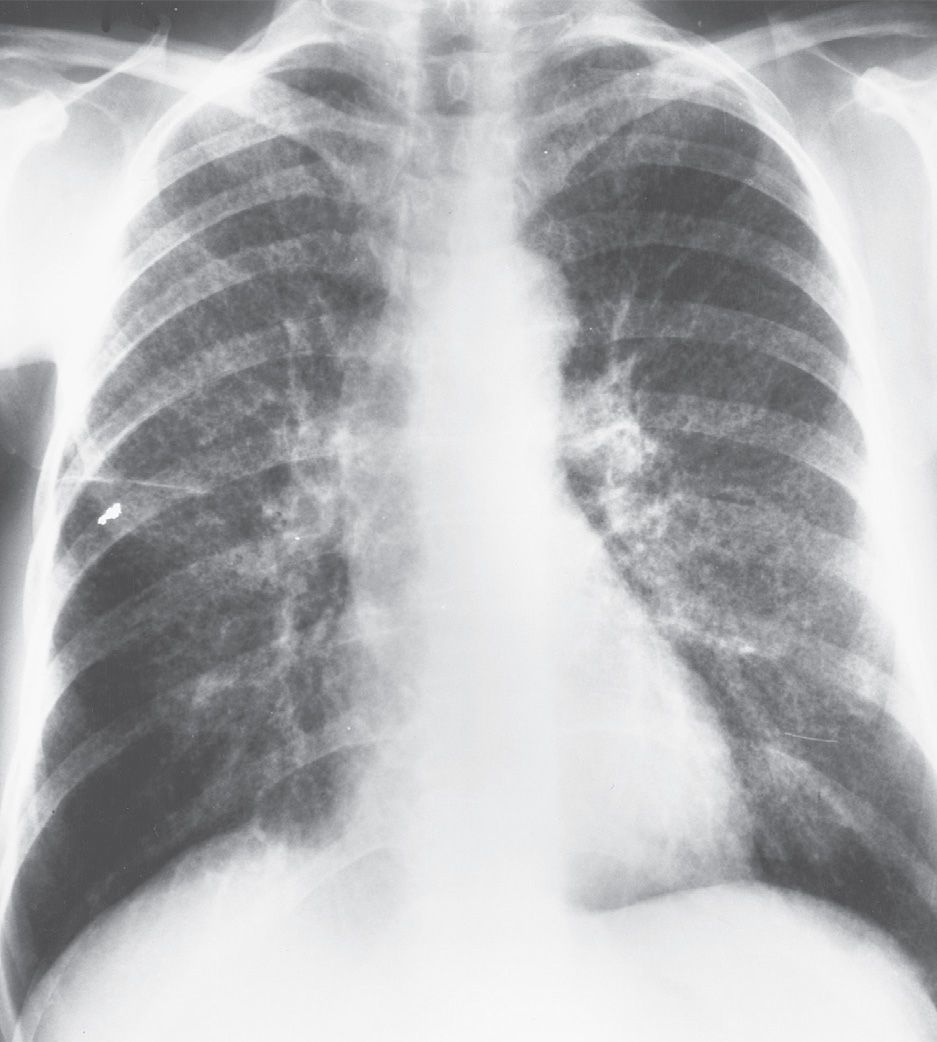
FIG. 10.5 • Sarcoidosis. PA chest radiograph shows reticulonodular opacities scattered throughout the upper and middle lungs. Parenchymal disease without lymphadenopathy indicates class III sarcoidosis.
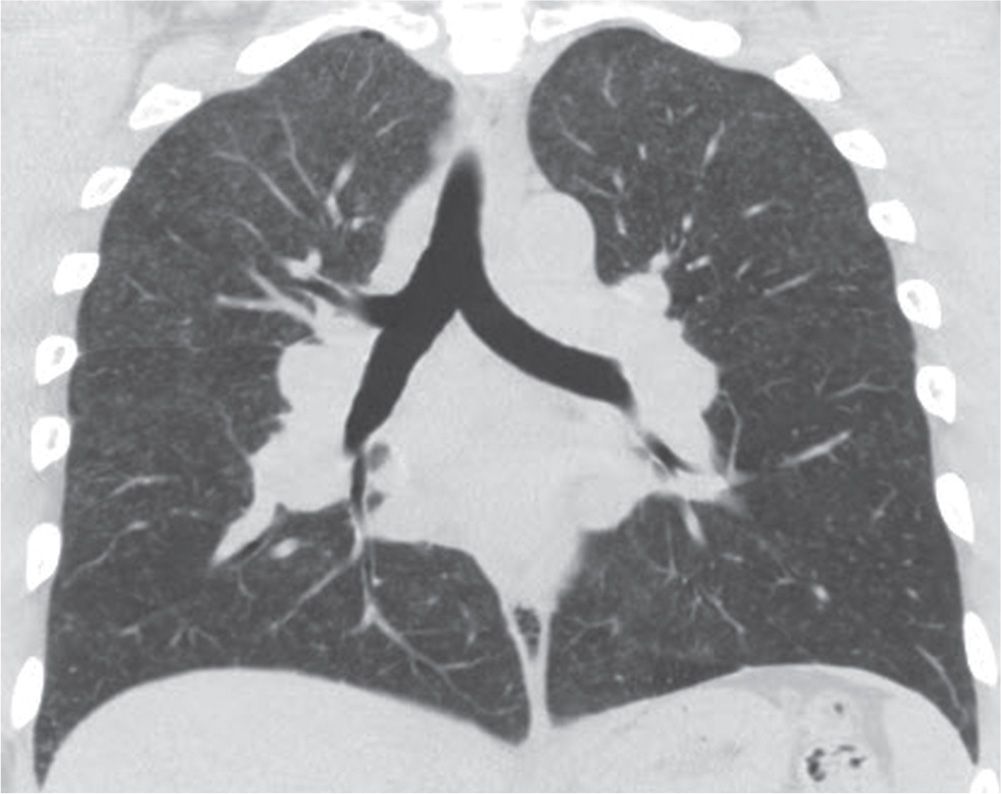
FIG. 10.6 • Sarcoidosis. Coronal CT shows multiple small nodules in a predominantly upper and middle lung distribution.
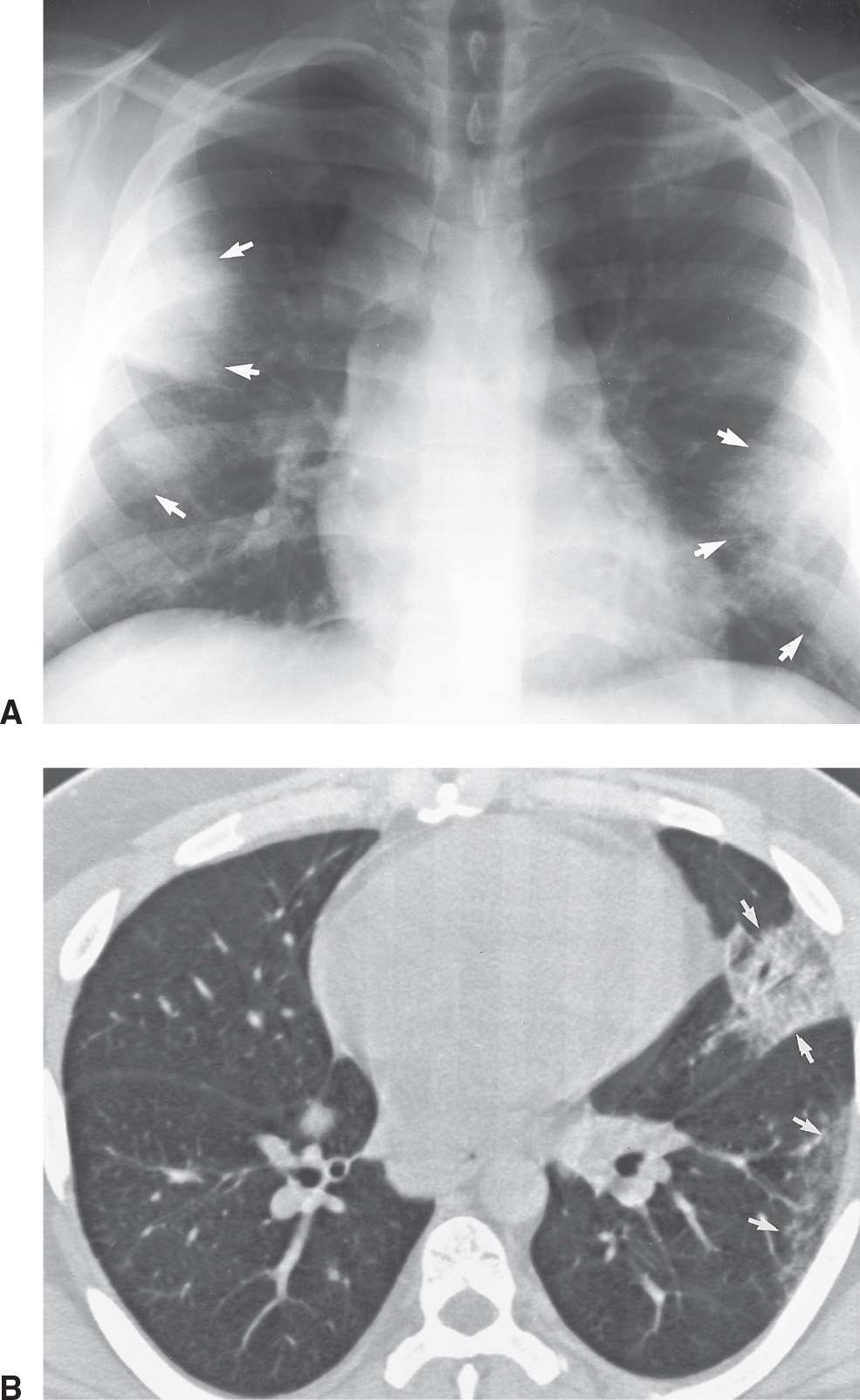
FIG. 10.7 • Sarcoidosis. A: PA chest radiograph of a 28-year-old man with mild shortness of breath shows right paratracheal and bilateral hilar lymphadenopathy and bilateral peripheral areas of consolidation (arrows). B: CT shows multifocal opacities in the periphery of the left lung (arrows). This pattern of sarcoidosis is referred to as alveolar sarcoid, although pathologically it is seen to be an interstitial process.
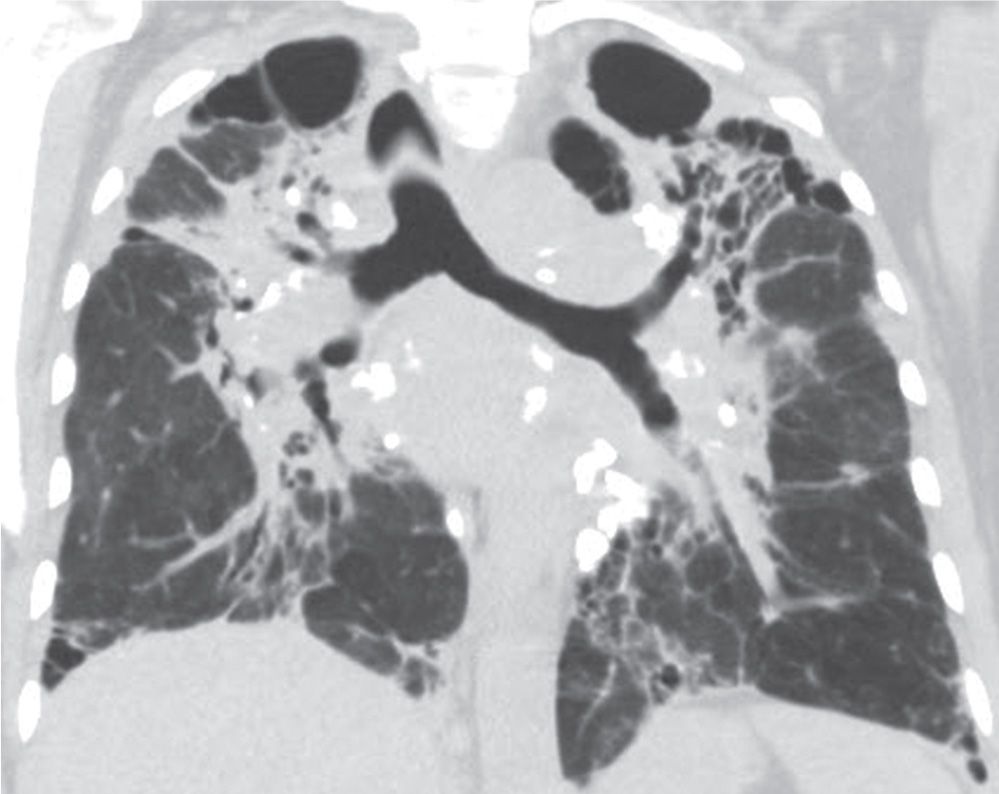
FIG. 10.8 • Sarcoidosis. Coronal CT shows bilateral upper lung–predominant fibrosis (class IV) with associated traction bronchiectasis, architectural distortion, upward retraction of the hila, and multiple calcified lymph nodes.
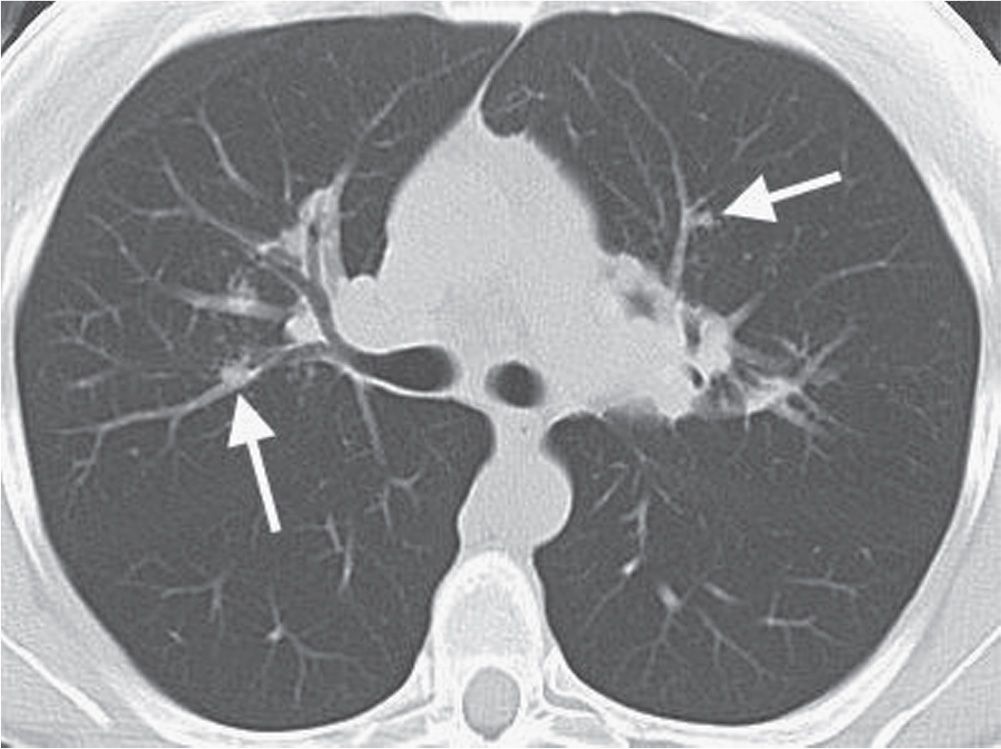
FIG. 10.9 • Sarcoidosis. CT of a 40-year-old man shows ill-defined nodules in a bronchovascular distribution (arrows).
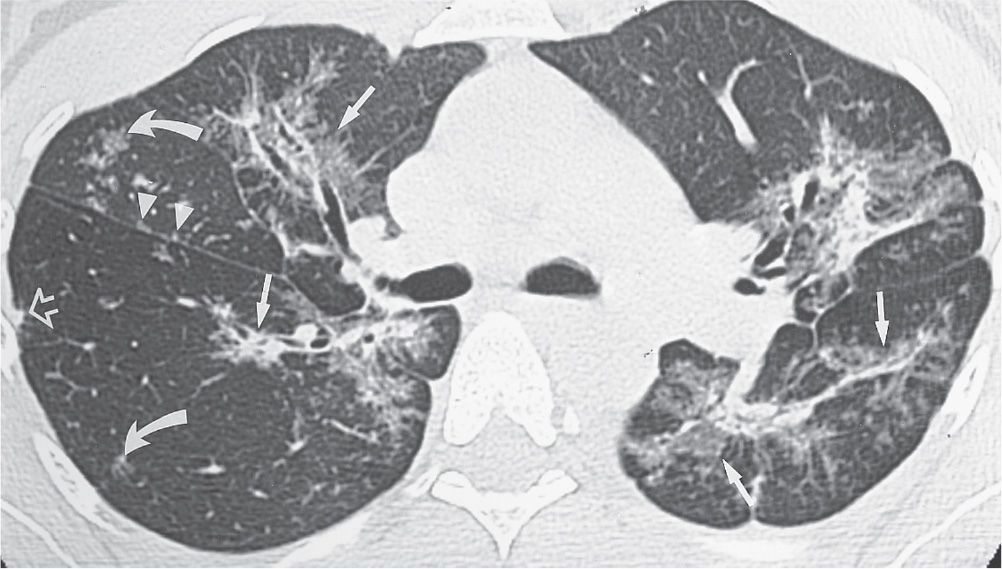
FIG. 10.10 • Sarcoidosis. CT image of a 46-year-old woman with mild shortness of breath shows bilateral areas of abnormal opacification distributed along the central and peripheral bronchovascular bundles (straight arrows). Some of the opacities are of ground-glass attenuation, allowing visualization of underlying bronchial and vascular markings. Note the small nodules along the right major fissure (arrowheads), in the center of secondary pulmonary lobules (curved arrows), and in a peripheral subpleural location (open arrow). All of these findings illustrate a perilymphatic distribution, which is typical of sarcoidosis.
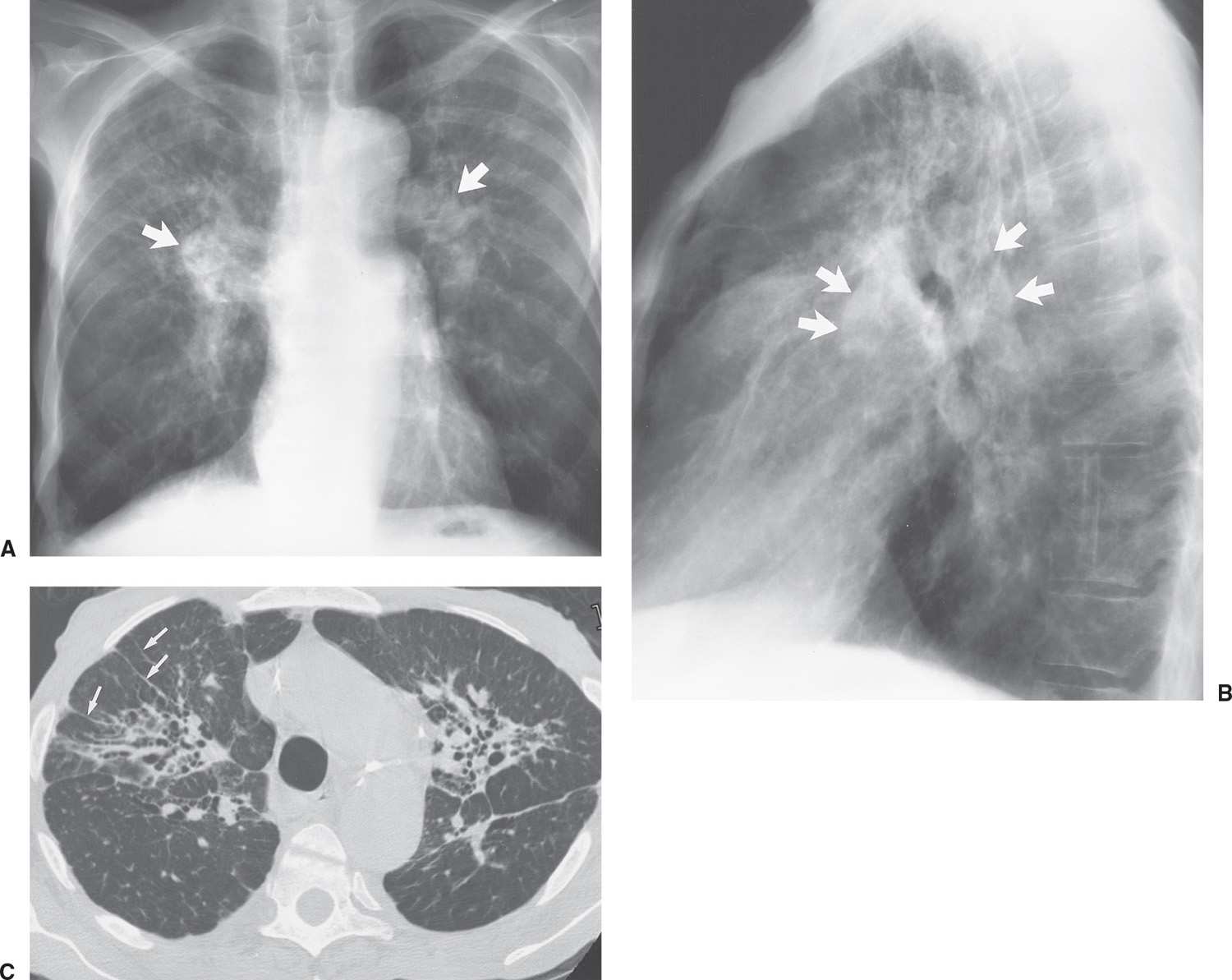
FIG. 10.11 • Sarcoidosis. PA (A) and lateral (B) chest radiographs of a 69-year-old man with class IV sarcoidosis show enlarged pulmonary arteries (arrows) secondary to pulmonary arterial hypertension. Note the diffuse upper and middle lung reticulonodular opacities, and note also the upward retraction of the hila. C: CT shows bilateral central areas of pulmonary fibrosis with thickening of bronchovascular bundles and traction bronchiectasis. Note the parenchymal bands on the right (arrows).
SILICOSIS
Silicosis is a disease of the lungs caused by inhalation of dust containing silicon dioxide, or silica, the predominant constituent of the earth’s crust. Silica dust is prevalent in mining, quarrying, and tunneling operations. Occupations associated with the development of silicosis include mining of heavy metals, the pottery industry, sandblasting, foundry work, and stonemasonry. When silica particles are inhaled, they are deposited in the alveoli and engulfed by alveolar macrophages, where they are acted on by lysosomal enzymes. The affected macrophage dies and liberates mediators (leading to stimulation of collagen production) and the silica particles. The silica particles are then free to be taken up by other macrophages, and the cycle continues, leading to progressive lung disease even without continued occupational exposure to silica. Silicosis can be classified as simple silicosis, complicated silicosis, acute silicosis, or Caplan syndrome. Coal worker’s pneumoconiosis, caused by inhalation of coal dust, is different pathologically from silicosis, but it produces chest radiographic findings similar to and often indistinguishable from those of silicosis.
Patients with simple silicosis are usually asymptomatic. Between 10 and 20 years’ exposure is usually necessary before the chest radiograph becomes abnormal (16). The chest radiograph shows multiple nodules, 1 to 10 mm in diameter, with a diffuse but upper lung–predominant distribution (Fig. 10.12). Occasionally, the nodules may calcify. Enlargement of mediastinal and hilar nodes is common and is occasionally associated with eggshell calcification similar to that seen with sarcoidosis.
Complicated silicosis refers to progression of simple silicosis, where the nodules become confluent and larger than 1 cm. On chest radiography, these opacities are seen predominantly in the periphery of the upper lungs, which, over time, tend to migrate or retract toward the hilum as the fibrotic process progresses. These conglomerate masses, which can reach several centimeters in size and contain obliterated blood vessels and bronchi, are referred to as progressive massive fibrosis (Figs. 10.13 and 10.14). The conglomerate masses are often surrounded by paracicatricial emphysema, which is best appreciated on chest CT. As conglomeration of the nodules occurs, the lungs gradually lose volume, and cavitation of the masses can occur. Patients who have advanced to this stage are at increased risk of active TB, and this diagnosis should be suspected when a new area of cavitation is seen on chest radiography.
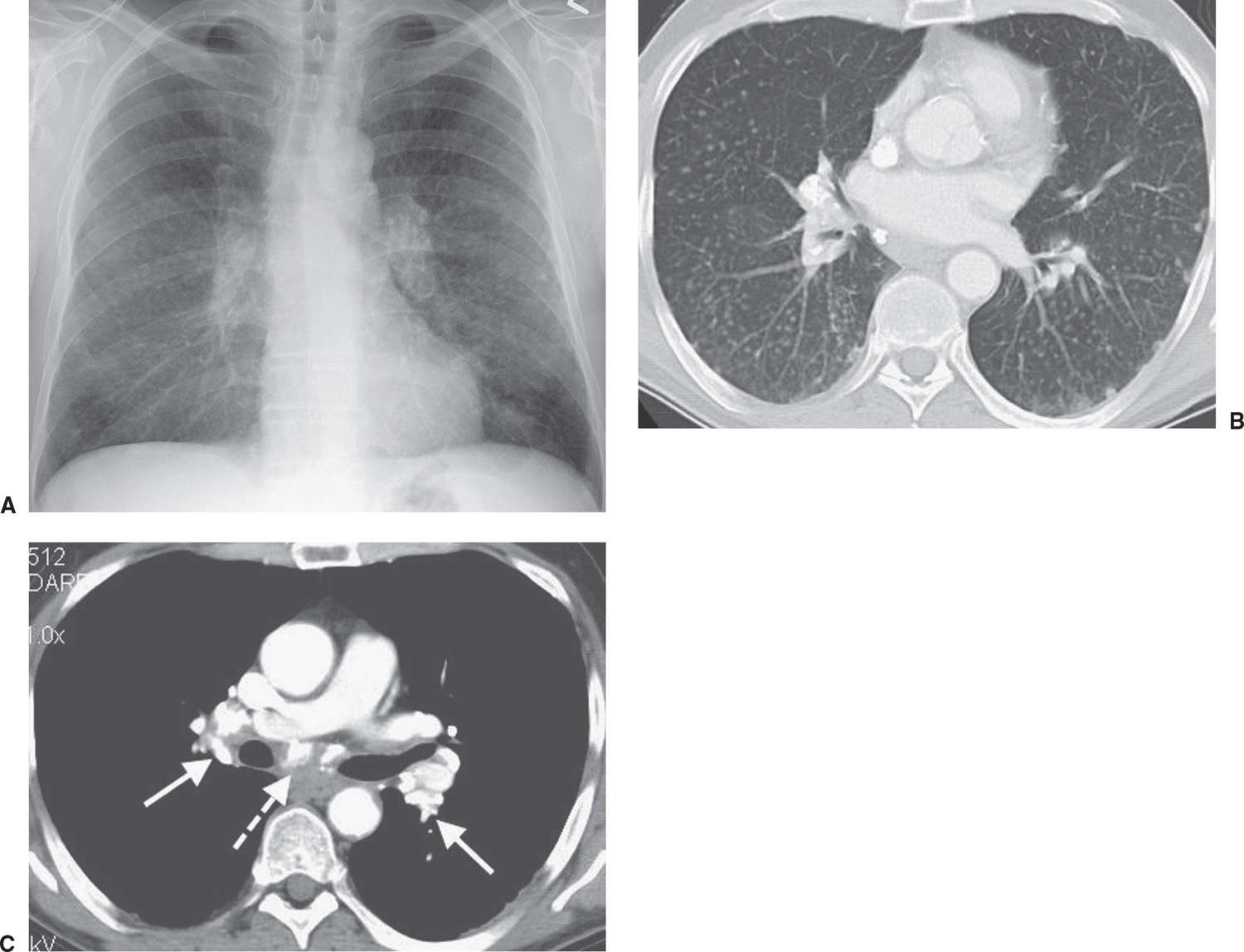
FIG. 10.12 • Simple silicosis. A: PA chest radiograph of a foundry worker shows numerous bilateral ill-defined tiny nodules, creating an overall increase in lung opacity. B: On CT, the nodules are much better appreciated. C: CT with mediastinal windowing shows densely calcified hilar (solid arrows) and subcarinal (dashed arrow) lymph nodes.
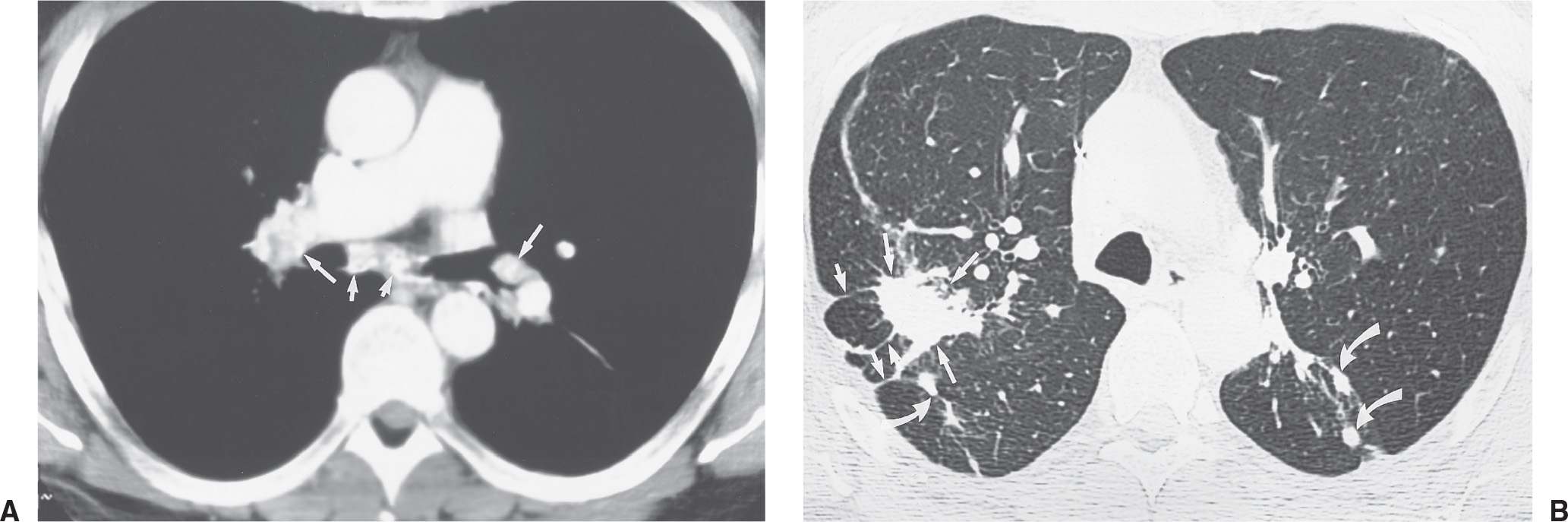
FIG. 10.13 • Complicated silicosis. A: CT of a 52-year-old man who spent many years working in a sand pit shows calcification of hilar (long arrows) and subcarinal (short arrows) nodes. B: CT with lung windowing shows “progressive massive fibrosis” in the right upper lobe (long straight arrows) and early conglomeration of nodules in the superior segments of the lower lobes (curved arrows). Multiple parenchymal bands are seen on the right (short straight arrows).
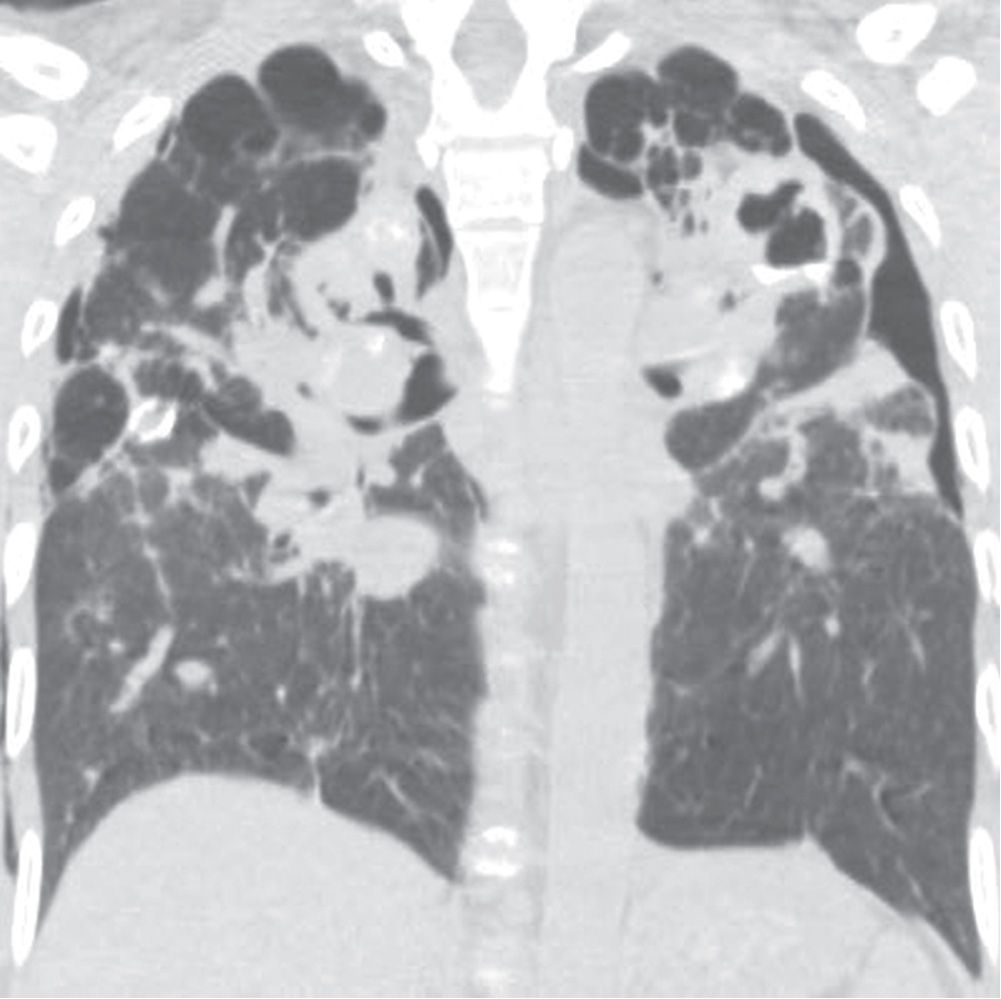
FIG. 10.14 • Complicated silicosis. Coronal CT shows bilateral upper lung–predominant fibrosis (“progressive massive fibrosis”), traction bronchiectasis, peripheral bullae, and parenchymal calcifications. The appearance is similar to that of class IV sarcoidosis.
Acute silicosis is a rare condition related to heavy acute exposure to silica in enclosed spaces with minimal or no protection. Histologically, the appearance is identical to that of pulmonary alveolar proteinosis; hence the term silicoproteinosis is used. The disease is rapidly progressive, often leading to death as a result of respiratory failure. The chest radiographic pattern is that of nonspecific diffuse airspace disease or ground-glass opacities, with a perihilar distribution and air bronchograms identical to the radiographic findings of pulmonary edema.
Caplan syndrome consists of the presence of large necrobiotic rheumatoid nodules superimposed on a background of simple silicosis. The syndrome is a manifestation of rheumatoid lung disease and is seen in both coal worker’s pneumoconiosis and silicosis.
LANGERHANS CELL HISTIOCYTOSIS
Langerhans cell histiocytosis (LCH), also referred to as histiocytosis X or eosinophilic granuloma, is a granulomatous disorder of unknown cause characterized by the presence within the granulomas of a histiocyte, the Langerhans cell. The disease is equally prevalent in male and female patients, is unusual in African-Americans (17), and 95% of adult patients are cigarette smokers (17). In most patients, symptoms appear in the third or fourth decade, but the disease can occur in teenagers and those over age 60. The diagnosis can be made in asymptomatic patients with abnormal chest radiographs. Pneumothorax is a classic manifestation of LCH, and the frequency of pneumothorax as the initial manifestation is as high as 14% (18). The pneumothoraces are commonly recurrent and may be bilateral. Approximately one-third of patients with LCH improve, one-third remain stable, and one-third deteriorate (18).
The chest radiograph of patients with LCH shows a diffuse, symmetric, reticulonodular pattern or, less commonly, a solely nodular pattern. Both patterns have a predominantly middle and upper lung distribution. The nodules are usually ill-defined, vary in size from 1 to 15 mm, and are often innumerable but can be few in number. Large nodules can mimic metastases. With time, small cystic airspaces develop, and larger airspaces up to 5 cm in diameter will form only rarely. Because of the development of these abnormal airspaces, lung volume does not decrease with time but often increases. Pleural effusion and hilar or mediastinal nodal enlargement are uncommon.
CT of the lungs shows cysts and nodules, often in combination (19,20) (Figs. 10.15 and 10.16). Cysts range in diameter from 1 to 30 mm or more and, unlike centrilobular emphysema, usually have very thin discrete walls and no centrilobular core structure. Some cysts have bizarre shapes, which along with an upper lung–predominant distribution can help to distinguish them from the uniformly round cysts that are typical of lymphangioleiomyomatosis. Nodules are typically 1 to 5 mm in diameter, have irregular margins, and may be cavitary. The disease is thought to progress from solid nodules to cavitary nodules to cysts, although this is controversial. End-stage disease can resemble that of generalized centrilobular pulmonary emphysema. In addition, as this disease is common in smokers, there is often superimposed centrilobular pulmonary emphysema.
RADIATION PNEUMONITIS
Radiation injury to the lung is most commonly seen after radiation therapy for breast cancer, lung cancer, and Hodgkin disease. On the chest radiograph, the changes of radiation pneumonitis are almost always confined to the field of irradiation. The first change is a diffuse haze in the irradiated region, with obscuration of the normal vascular markings. Patchy opacities appear, which may coalesce into a nonanatomic but geometric area of pulmonary opacification. These radiographic changes usually appear about 8 weeks after treatment, depending on the radiation dose and dosing interval (21); peak reaction occurs at 3 to 4 months. With time, the opacities become more linear or reticular, and fibrous contraction and distortion of lung architecture occurs. The fibrosis and contraction continue over a 12- to 18-month period. When only the apices of the lung are affected by radiation, such as with treatment for head and neck neoplasms, the radiographic changes do not appear geometric but ill-defined and patchy (Fig. 10.17). When bilateral apical airspace opacities are seen on chest radiography, radiation pneumonitis should be considered in the appropriate patient population. Radiation of the axilla, an adjuvant treatment for patients with breast cancer who have undergone lumpectomy or mastectomy, can result in ipsilateral peripheral upper lung patchy airspace opacities (Fig. 10.18).
TUBERCULOSIS
Once a disease of childhood, more than half of cases of initial infection with Mycobacterium tuberculosis, or primary TB, are now seen in the adult population (22). In primary tuberculous infections, the pulmonary focus and lymphadenopathy may resolve without a trace, or they may leave a focus of caseous necrosis, scarring, or calcification. Several terms are used to describe the form of TB that develops after a primary infection under the influence of established hypersensitivity including reactivation TB, postprimary TB, and secondary TB.
The predominant radiographic feature of primary TB is the presence of hilar lymphadenopathy (usually unilateral) and mediastinal lymphadenopathy contiguous to the affected hilum. Lymphadenopathy is less common and milder in adults than in children, with the exception of immunocompromised adults, especially those with AIDS. On CT, the enlarged nodes typically have a low-density center with rim enhancement (Fig. 10.19) (23). The pulmonary foci of primary TB are randomly distributed throughout the lungs, and they range from small, occasionally imperceptible, ill-defined parenchymal opacities to segmental or lobar consolidation, often with an appearance similar to that of other bacterial pneumonias (Figs. 10.20 and 10.21). The incidence of cavitation varies between 10% and 30% (22). Hilar or mediastinal lymph node calcification is observed in 35% of cases (24). Pleural effusions are not uncommon, are generally unilateral, and are usually, but not always, associated with some identifiable pulmonary parenchymal disease (Fig. 10.22). Bronchial stenosis, bronchial occlusions, and polypoid endobronchial tuberculous lesions may be seen on CT. It should be noted that in some immunocompromised patients, their “reactivation” TB presents clinically and radiographically like primary infection seen in immunocompetent patients.
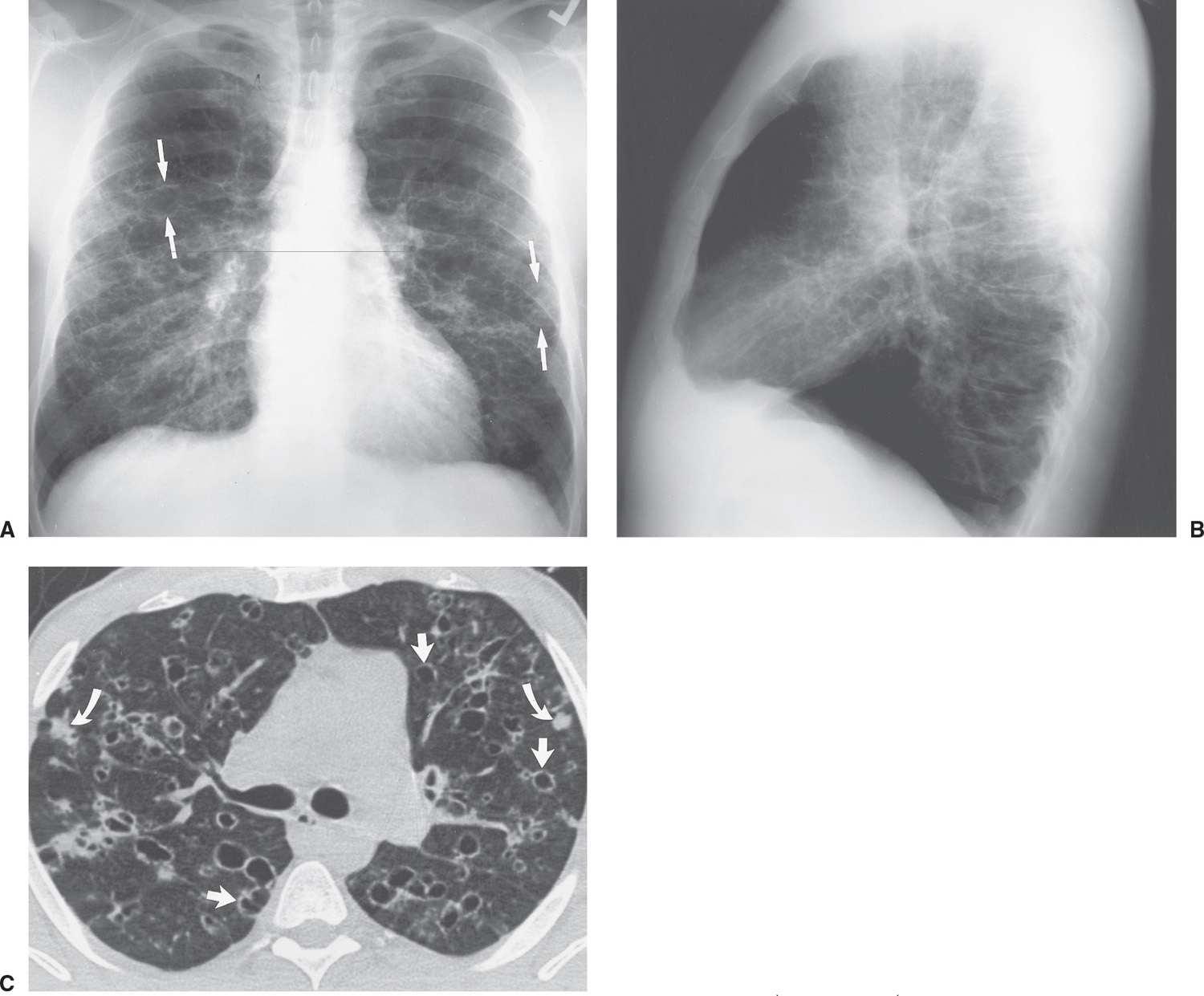
FIG. 10.15 • Langerhans cell histiocytosis. PA (A) and lateral (B) chest radiographs of a 32-year-old male cigarette smoker show bilateral reticular interstitial opacities and thin-walled cysts (arrows). Note the increased lung volumes. C: CT shows bilateral thin-walled cysts, with rounded and irregular shapes (straight arrows), and ill-defined nodules (curved arrows).
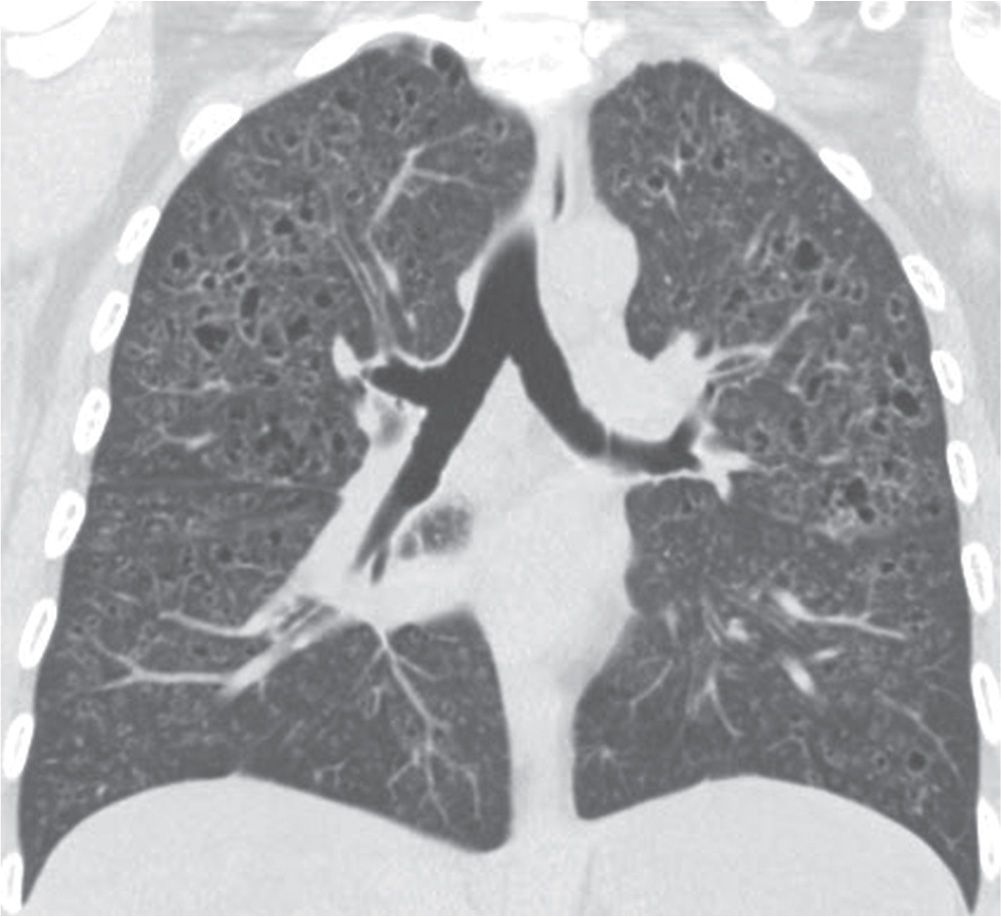
FIG. 10.16 • Langerhans cell histiocytosis. Coronal CT shows numerous small, irregular cysts with defined walls in an upper lung–predominant distribution.
The earliest chest radiographic findings of reactivation TB consist of one or more ill-defined patchy opacities, with or without small satellite foci in the adjacent lung, occurring in the posterior segments of the upper lobes in the majority of patients and in the superior segment of the lower lobes in most of the remainder of patients. This distribution of disease is very helpful in suggesting the diagnosis of reactivation TB. Cavitation with or without the presence of air–fluid levels is a distinct feature of reactivation TB and indicates a high likelihood of active infection (Figs. 10.23 to 10.27). Endobronchial spread of disease, with “tree-in-bud” opacities, best seen with CT, also suggests active infection. In the proper clinical setting, the presence of cavitation and endobronchial spread implies that the disease is highly contagious, and patients with cavitary disease should be placed under immediate infective precautions (respiratory isolation) on the basis of radiographic findings alone. With healing, the chest radiograph shows gradually increasing definition of the lung opacities, development of fibrosis in the surrounding lung, contraction and volume loss of the affected segment or lobe with fissural displacement or distortion of the vascular structures in the hilum, bronchiectasis, and calcification (Fig. 10.28). Fluid levels in cavities disappear, and the cavities either disappear or persist with a smooth inner wall. It should be noted that patients can rarely still have sputum-positive TB and a normal chest radiograph.
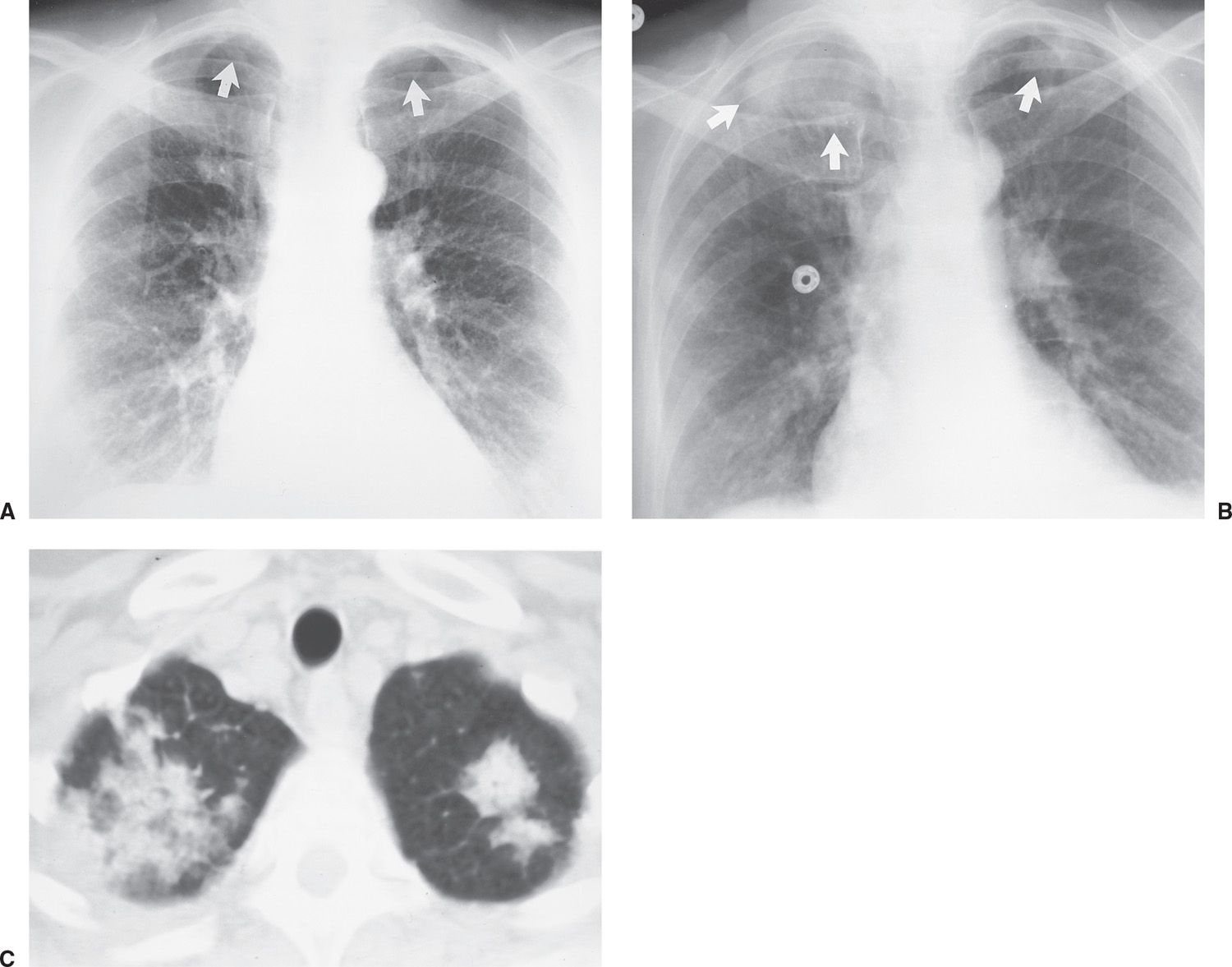
FIG. 10.17 • Radiation pneumonitis. A: PA chest radiograph of a 60-year-old woman 3 months after radiation treatment to the neck for a piriform sinus carcinoma shows subtle areas of abnormal opacification in both apices (arrows). B: PA chest radiograph obtained 2 months later shows progression of apical opacities (arrows). C: CT shows bilateral apical airspace disease without anatomic or geographic distribution.
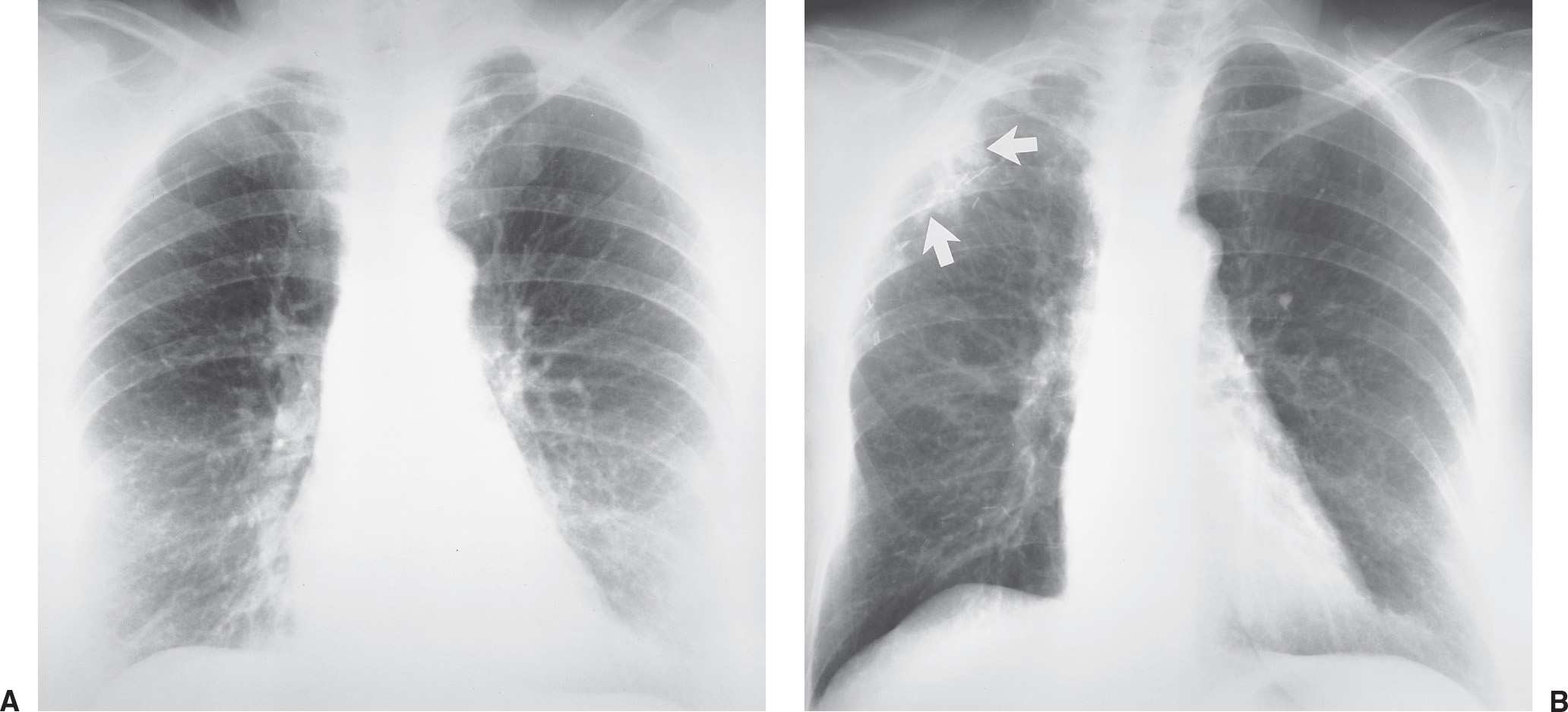
FIG. 10.18 • Radiation pneumonitis. A: Normal baseline PA chest radiograph of a 71-year-old woman. B: PA chest radiograph obtained 11 months later shows interval right mastectomy for treatment of breast cancer (note the hyperlucent right lower hemithorax) and surgical clips in the right axilla. Note new abnormal areas of nonsegmental opacification in the periphery of the right upper lobe (arrows) from recent radiation to the axilla.
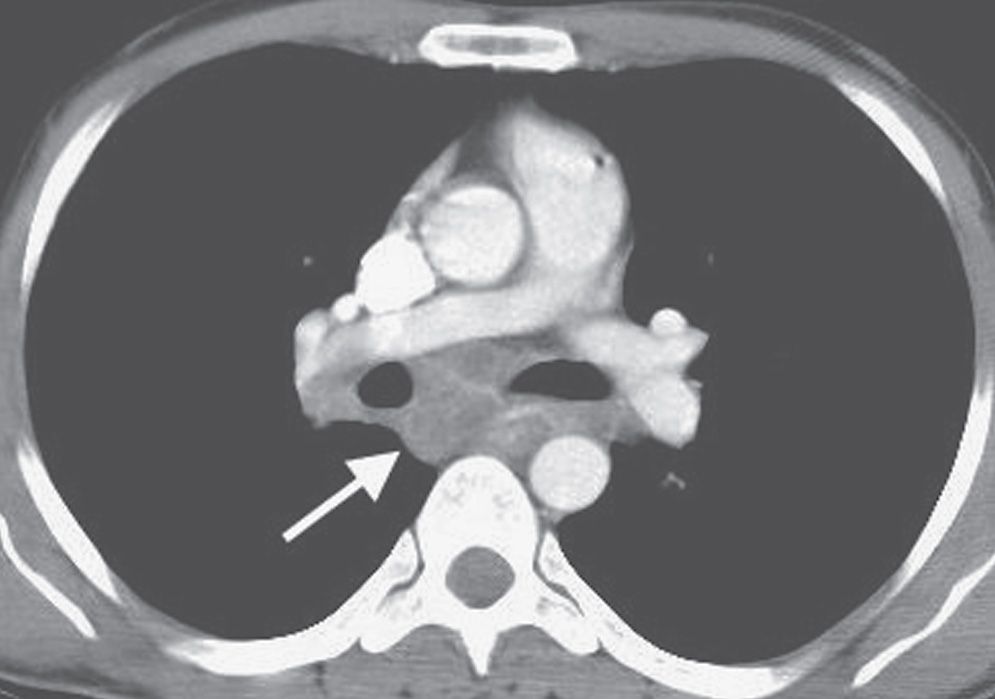
FIG. 10.19 • Primary tuberculosis. CT shows low-density subcarinal nodes with partial rim enhancement (arrow).
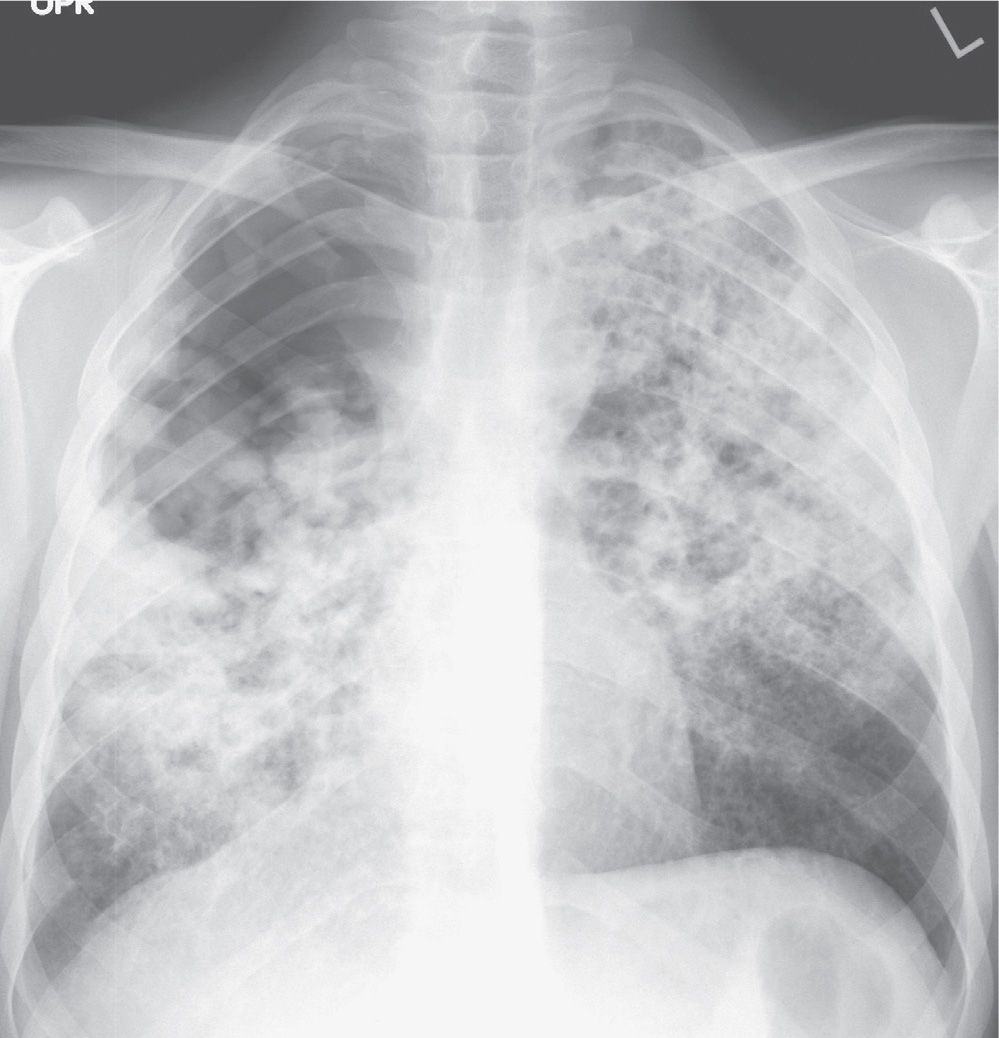
FIG. 10.20 • Primary tuberculosis. PA chest radiograph shows diffuse nodular airspace disease.
Other patterns of reactivation TB include lobar pneumonia, diffuse bronchopneumonia, endobronchial TB, tuberculoma formation, miliary TB, and tuberculous pleuritis (Fig. 10.29). A calcified lymph node may erode into an adjacent airway, becoming a broncholith, and be associated with hemoptysis or postobstructive atelectasis or pneumonia. Broncholiths can be suggested when a previously documented nodal calcification on chest radiography has disappeared or changed position. On rare occasion, a patient can cough up pieces of a calcified broncholith, a phenomenon referred to as lithoptysis. Tuberculomas are discrete tumorlike foci of TB in which there is a fine balance between inflammation and healing. The margins of a tuberculoma are usually well-circumscribed. Tuberculomas may be single or multiple, are occasionally as large as 5 cm in diameter, and may grow slowly over an extended period of time. Calcification develops in the central caseous core with time and may be seen radiographically, but it is better characterized with CT. When the calcification is dense and assumes the majority of the volume of the tuberculoma as seen on chest radiography, the diagnosis of a benign inactive granuloma can be assumed. Such a parenchymal tuberculoma, known as a Ghon lesion, in combination with calcified nodes, is referred to as a Ranke complex. Miliary TB, which results from hematogenous dissemination of disease, is an uncommon but serious complication of both primary and reactivation TB. The chest radiograph shows innumerable 2- to 3-mm nodules likened to millet seeds in size and appearance. The nodules are uniformly distributed and equal in size. Because there is a threshold below which the nodules are imperceptible, miliary TB can be present in patients with a “normal” chest radiograph. Miliary TB does not leave residual calcifications.
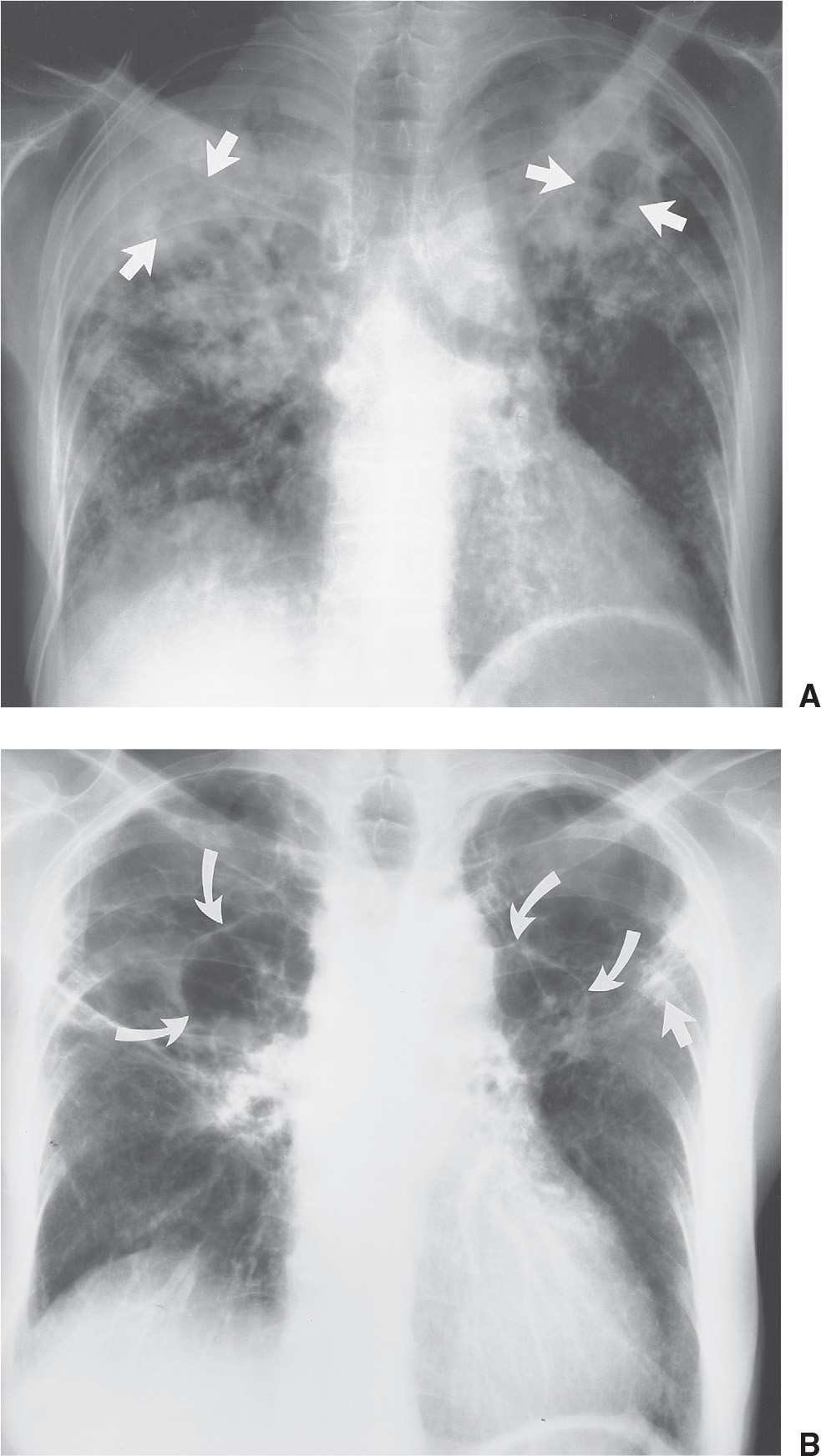
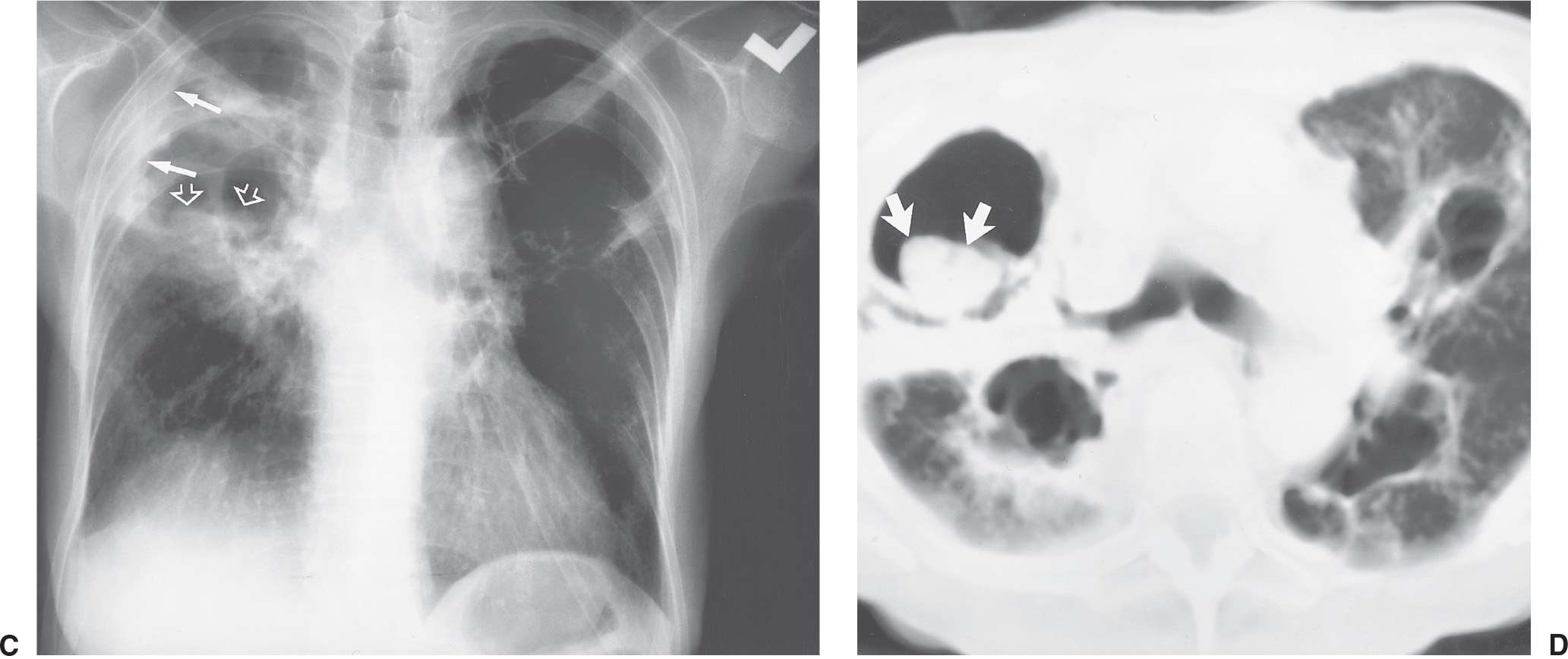
FIG. 10.21 • Primary tuberculosis. A: PA chest radiograph of a 71-year-old man with fever, hemoptysis, and weight loss shows bilateral patchy airspace opacities, with areas of cavitation in the upper lobes (arrows). Sputum contained numerous M. tuberculosis organisms. B: PA chest radiograph taken 9 months later shows changes of healing in upper lobes consisting of linear opacities (straight arrow) and thin-walled cavities (curved arrows). C: PA chest radiograph 2 months after (B) shows new right apical pleural thickening (solid arrows) and increased opacification of the right upper lobe. A fungus ball (aspergilloma) is seen within a right upper lobe cavity (open arrows). D: CT shows a fungus ball in the right upper lobe cavity (arrows). Note the cystic changes of healed TB in the left upper lobe.
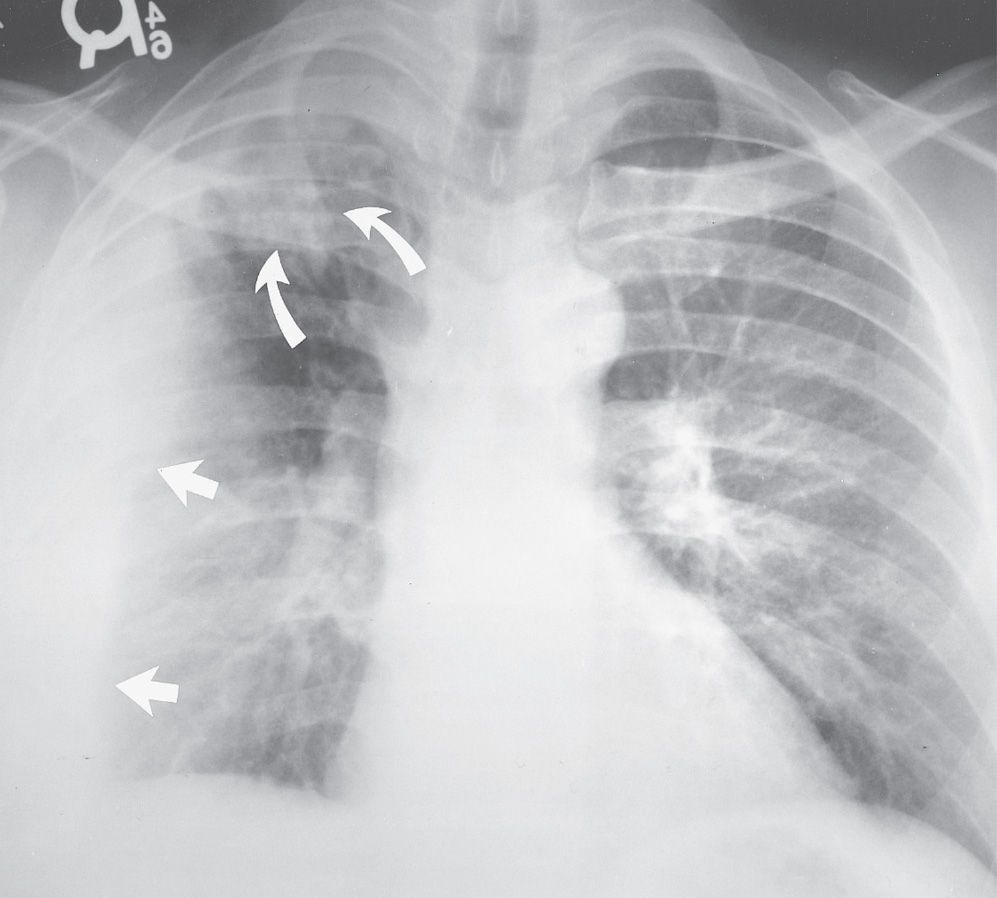
FIG. 10.22 • Primary tuberculosis. PA chest radiograph of a 39-year-old man shows abnormal right upper lobe opacification (curved arrows) and a large, loculated, right pleural effusion (straight arrows). Freely layering pleural fluid collects inferiorly, within the most gravity-dependent portion of the pleural space, in upright positioning, and the pleural fluid in this case tracks superiorly along the chest wall.
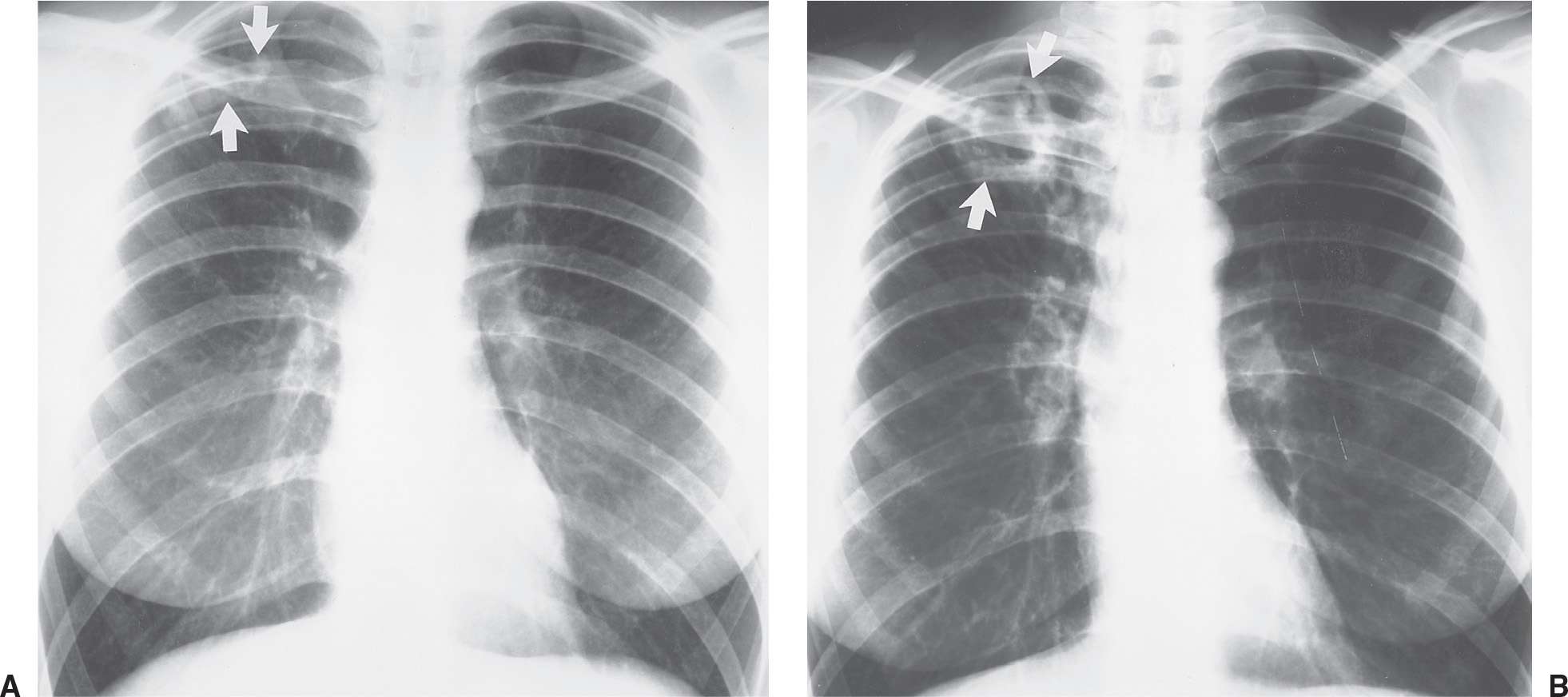
FIG. 10.23 • Reactivation tuberculosis. A: PA chest radiograph of a 44-year-old man shows a partially calcified opacity in the right upper lobe (arrows), which was unchanged in comparison with multiple prior chest radiographs and thus consistent with prior TB infection of indeterminate activity. B: PA chest radiograph obtained 6 years later shows a new cavity in the right upper lobe (arrows). Sputum contained M. tuberculosis organisms.
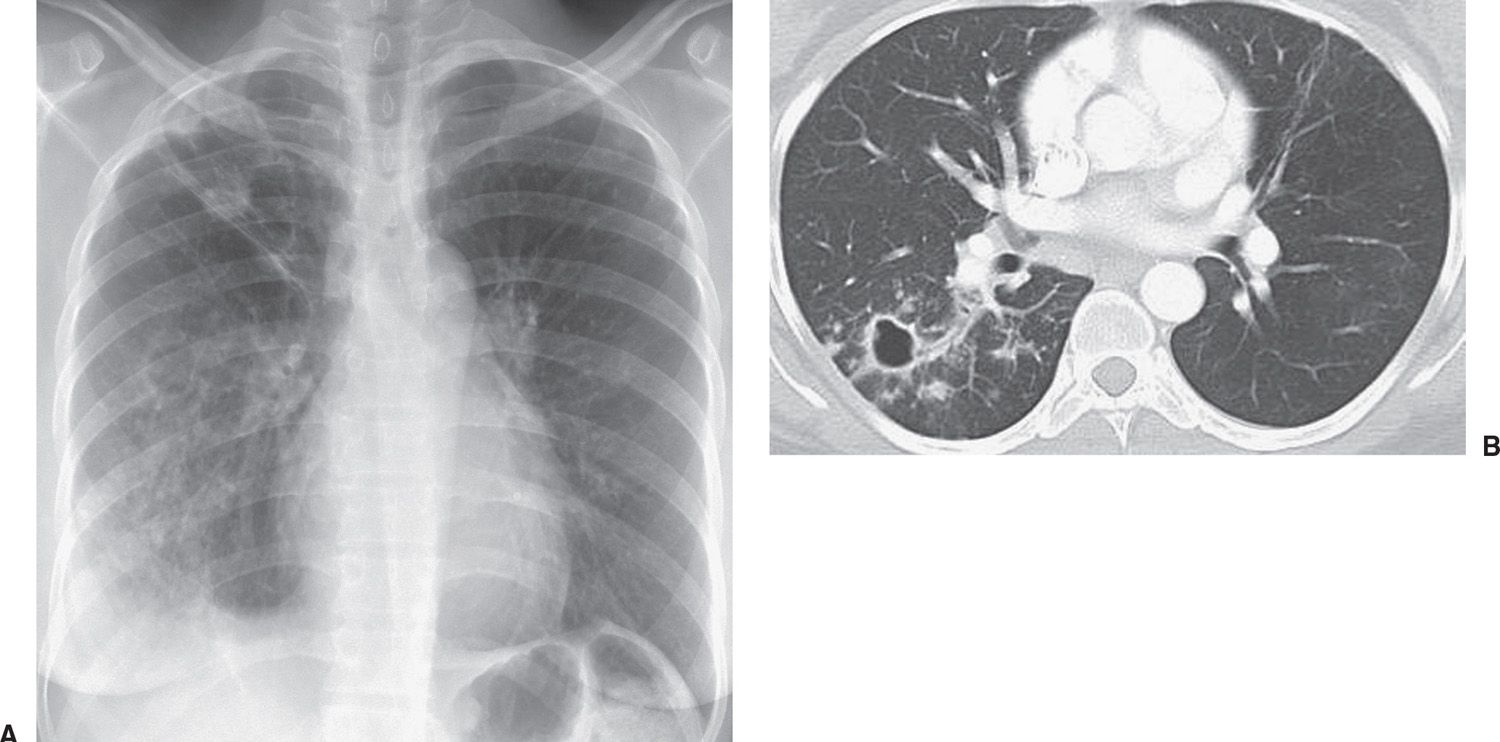
FIG. 10.24 • Reactivation tuberculosis. A: PA chest radiograph of a 30-year-old man from Africa who had been treated for TB 10 years earlier shows elevation of the minor fissure, right apical opacity, and ill-defined opacities throughout the right lung. B: CT shows a thin-walled cavity, thickening of the bronchovascular bundles, and ill-defined nodules in the superior segment of the right lower lobe.
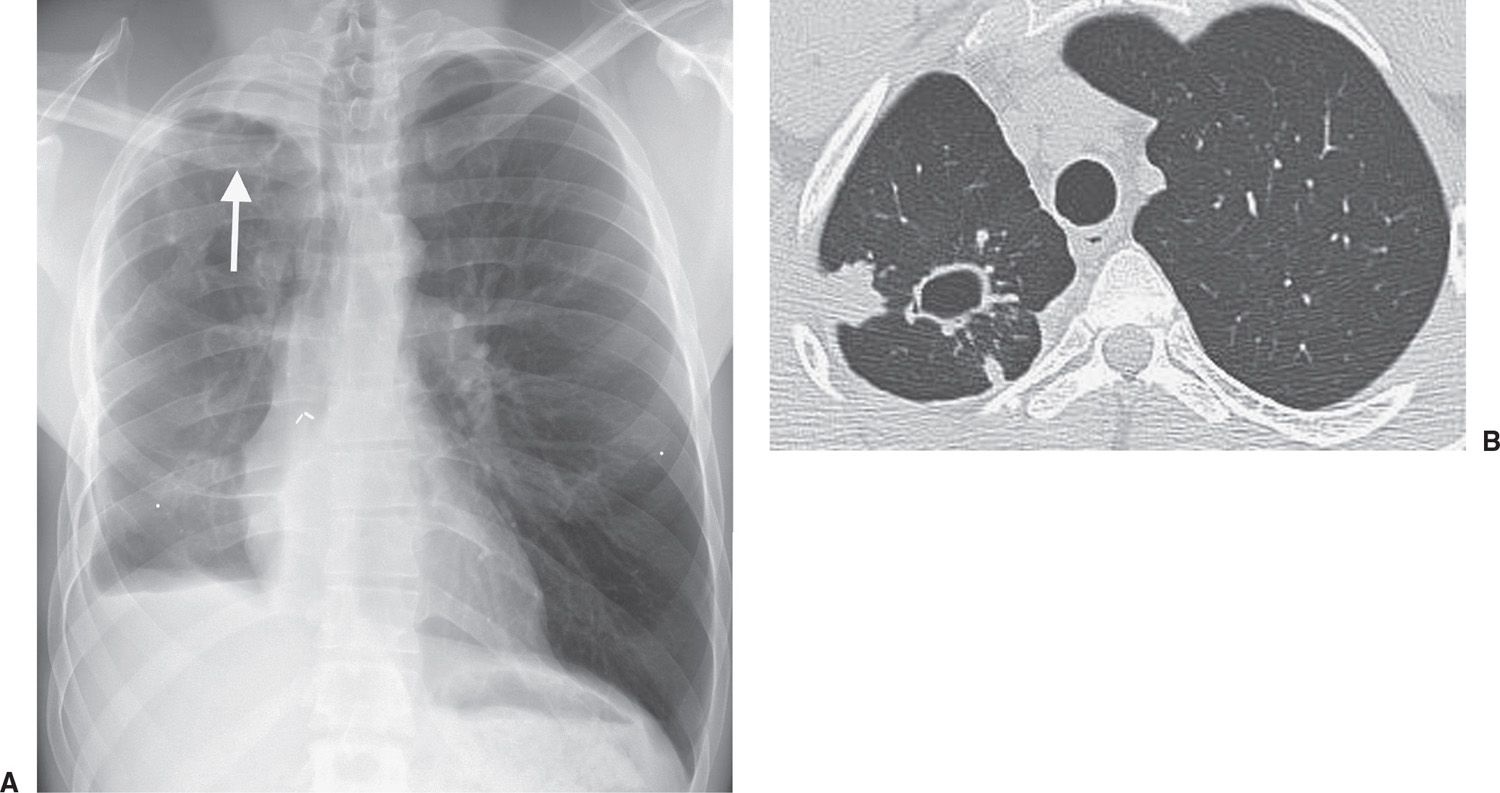
FIG. 10.25 • Reactivation tuberculosis. A: PA chest radiograph of a 28-year-old man with a prior history of right middle and lower lobectomy and right pleurodesis, currently taking steroids for severe asthma, shows right apical opacity and a thin-walled cyst (arrow) in the right upper lung. Both were new findings compared with prior chest radiographs. B: CT shows the cyst and surrounding ill-defined nodules in the posterior right upper lobe.
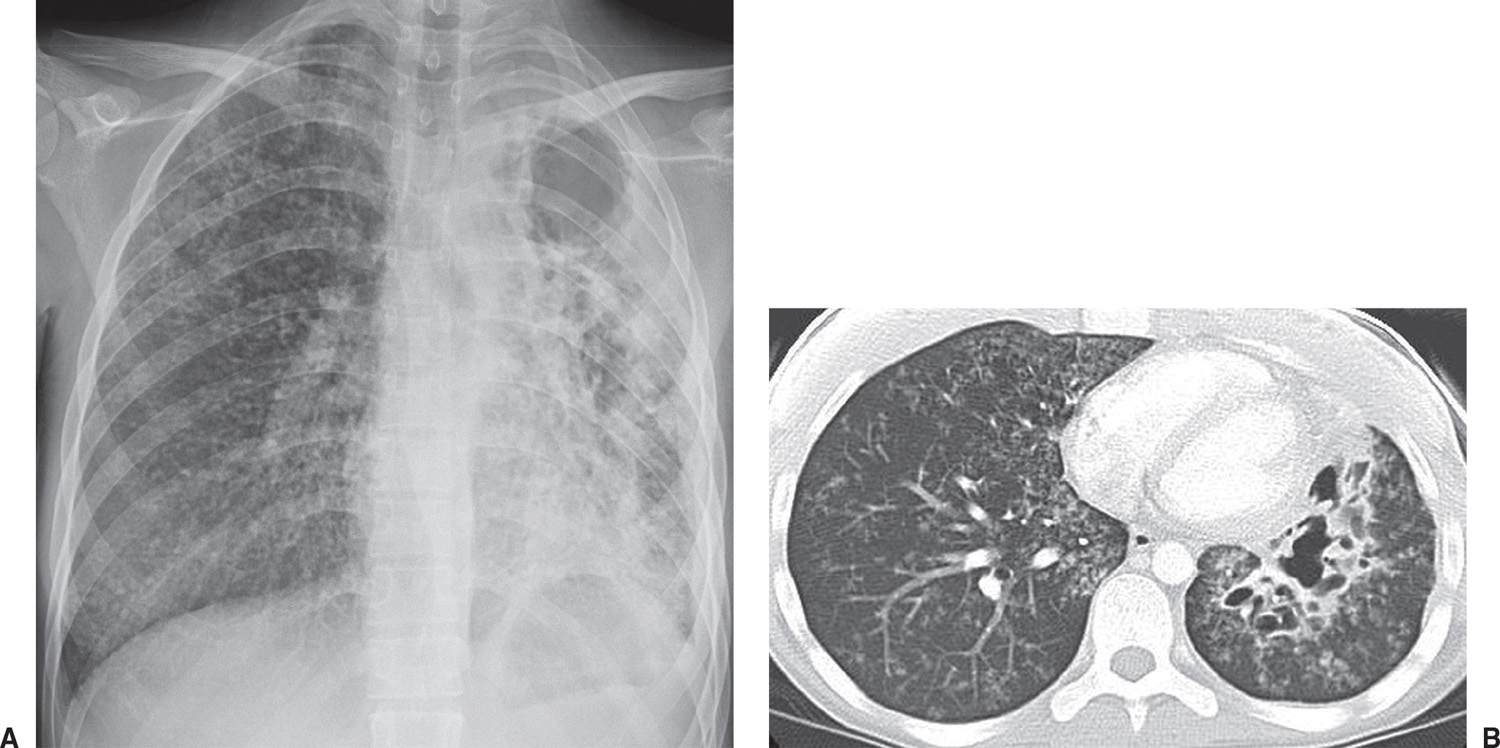
FIG. 10.26 • Tuberculosis. A: PA chest radiograph of an 18-year-old man with multidrug-resistant TB, cough, night sweats, and weight loss shows diffuse small nodules in the right lung, and cavitary disease on the left associated with volume loss. B: CT shows better the cavitary disease in the left lower lobe and bilateral small nodules.
A number of mycobacteria other than M. tuberculosis can cause pulmonary infection. These atypical mycobacteria are common in the natural environment, and numerous species exist, including Mycobacterium kansasii and Mycobacterium avium-intracellulare; the latter is a very important pathogen in human immunodeficiency virus (HIV)–infected individuals. The classic features of atypical mycobacterial infections of the lung are those of a chronic indolent fibrocavitary process, usually involving one or both apical regions of the lungs in a middle-aged or older individual with underlying chronic obstructive pulmonary disease (25), or in chronic lower lung bronchiectasis. In many cases, the radiographic features are indistinguishable from those of reactivation TB. The lesions have the same predilection for the posterior aspects of the upper lobes or the superior segments of the lower lobes as reactivation TB. Cavitation occurs in up to 96% of patients with atypical mycobacterial infection (Fig. 10.30) (26).
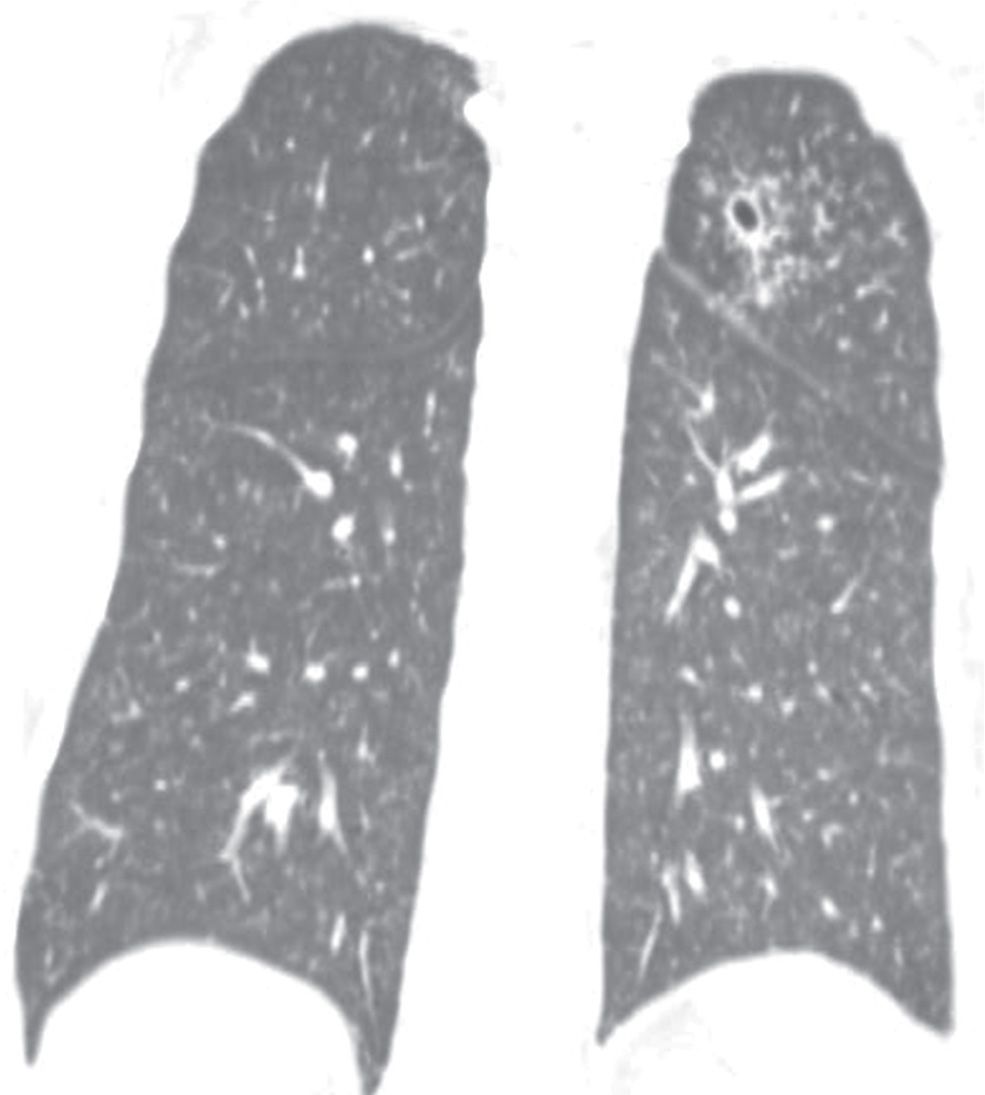
FIG. 10.27 • Tuberculosis. Coronal CT shows cavitary disease in the left upper lobe and bilateral diffuse miliary nodules.
ASPERGILLUS LUNG DISEASE
Fungi of the genus Aspergillus are ubiquitous saprophytes that are commonly laboratory contaminants but on occasion can become human pathogens. Infection is acquired primarily through the respiratory tract. Exposure to Aspergillus is almost universal, yet the nonimmunocompromised patient usually does not develop Aspergillus infection (27).
Aspergillosis represents a spectrum of diseases based on the degree of immune impairment at one end of the spectrum and hypersensitivity at the other end of the spectrum (28). Four basic types of pulmonary aspergillosis are (i) noninvasive (saprophytic), (ii) semi-invasive, (iii) invasive, and (iv) allergic.
Noninvasive pulmonary aspergillosis refers to colonization of a preexisting cavity or cystic area in the lung with Aspergillus organisms, creating a “fungus ball” or mycetoma. Prior tuberculous infection, emphysematous bullae, sarcoidosis (Fig. 10.31), or other processes, such as ankylosing spondylitis, that result in cavities, cysts, or cystic bronchiectasis, can lead to mycetoma formation (29,30). In noninvasive aspergillosis, the fungus causes the development of vascular granulation tissue within the cavity wall, which can lead to hemoptysis. With peripheral cavities, there is also a pleural response to the presence of the organism, leading to marked pleural thickening and extrapleural fatty hypertrophy as clues to the chronic inflammation. Chest radiography and CT typically show a mobile, intracavitary mass, usually in an upper lobe (Figs. 10.32 and 10.33). Specific therapy is not required for patients with an asymptomatic aspergilloma; however, this type of aspergillosis can cause massive hemoptysis that can sometimes lead to death. Bronchial artery embolization, surgery, or instillation of amphotericin B into the aspergilloma cavity can be used to treat patients with an aspergilloma and hemoptysis.
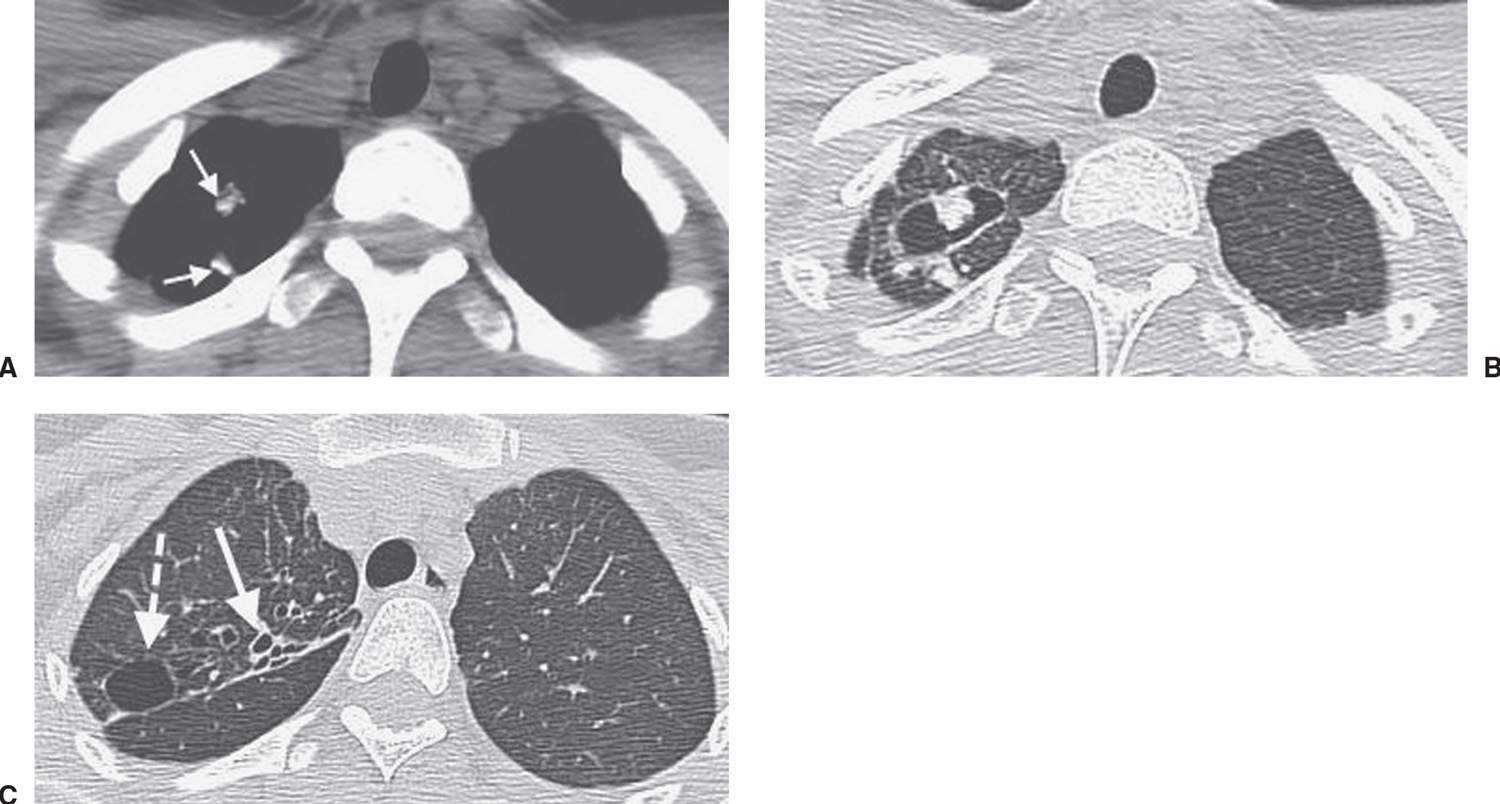
FIG. 10.28 • Healed tuberculosis. A: CT shows calcified nodules in the right upper lobe (arrows). B: CT with lung windowing shows a thin-walled cyst. The mural nodule along the anterior cyst wall and the nodule posterior to the cyst represent the calcified nodules seen in (A). C: CT at a more inferior level shows bronchiectasis (solid arrow) and a second thin-walled cyst (dashed arrow). The findings were unchanged in comparison with multiple CT scans obtained during the prior 3 years.
Semi-invasive pulmonary aspergillosis refers to a chronic, indolent form of Aspergillus infection that leads to cavity formation and then a classic aspergilloma. This form of aspergillosis tends to occur in patients with depressed immune systems (e.g., those with a neoplasm, those who have undergone radiation, those who are of advanced age or debilitated, those with diabetes or chronic obstructive lung disease, or those on chronic corticosteroid therapy) (31). The disease typically begins as an upper lung opacity on chest radiography, which, over a period of weeks to months, gradually develops cavitation with formation of an “air crescent sign.” As the disease progresses, extensive apical pleural thickening develops. As cavitation progresses, a thick-walled cavity often becomes thin-walled, containing a mycetoma (Fig. 10.34). Therapy is the same as for noninvasive aspergillosis.
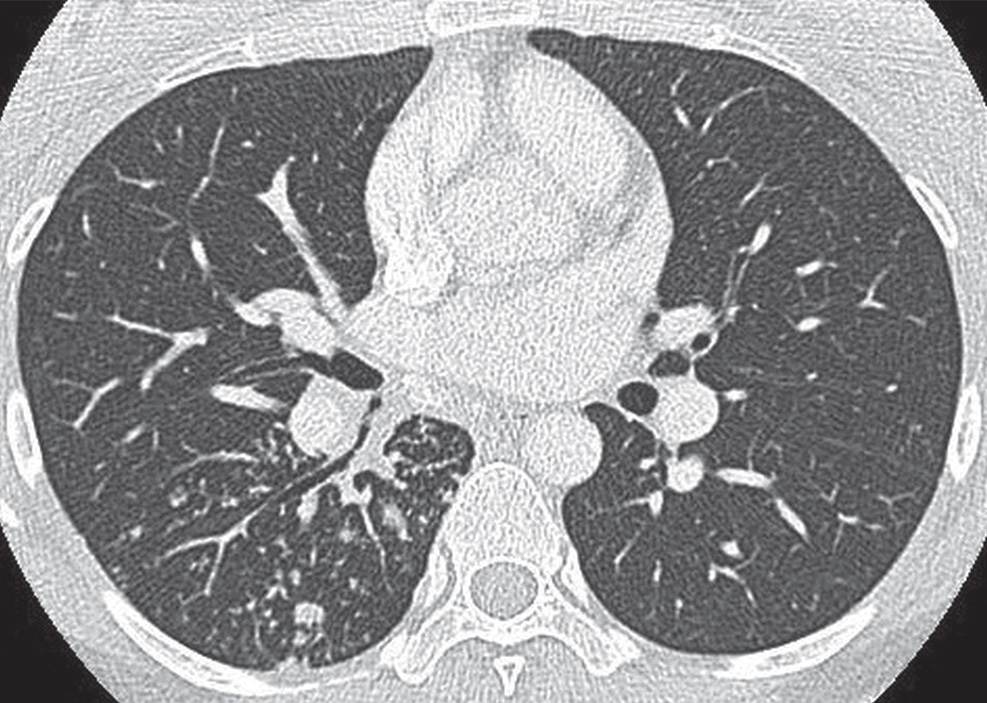
FIG. 10.29 • Reactivation tuberculosis. CT shows bronchiolar opacities in the superior segment of the right lower lobe, indicative of infectious disease.
Invasive pulmonary aspergillosis is the most pathologically aggressive form of aspergillosis and occurs in severely immunocompromised patients (Fig. 10.35). Chest radiographs show a solitary pulmonary nodule or mass, or multifocal opacities. In some cases, multiple poorly defined nodules (likely representing infarcts) are seen and extend to the periphery of the lungs. In patients with leukemia, cavitation of the nodules develops as the neutrophil count recovers from the chemotherapy-induced nadir (32). Cavitation leads to the air crescent sign, which is seen as a crescent-shaped collection of air between the cavity wall and the intracavitary contents. In contrast to noninvasive and semi-invasive forms of aspergillosis, in which a fungus ball occupies the cavity, in invasive aspergillosis the central mass within the cavity is almost always necrotic lung and not an aspergilloma. Early in the course of infection during bone marrow aplasia, before air crescent formation or cavitation, CT often shows the CT “halo sign,” a zone of ground-glass opacification surrounding the pulmonary nodule or mass. In patients with acute leukemia, the presence of the CT halo sign (representing a zone of hemorrhage) strongly suggests the diagnosis of invasive aspergillosis (Fig. 10.36) (33). Voriconazole is considered the drug of choice for invasive aspergillosis. Patients who do not respond to therapy usually die of the disease.
Allergic bronchopulmonary aspergillosis (ABPA) is a form of hypersensitivity reaction to inhaled Aspergillus organisms. It is seen (i) most commonly in patients with underlying asthma, (ii) in approximately 10% of patients with cystic fibrosis, and (iii) occasionally in patients with no known underlying pulmonary disease (34). The fungus grows noninvasively within the bronchi, releasing an antigen that causes host sensitization and a subsequent immunologic reaction. Mucus traps the organisms in the bronchi. Bronchiectasis either results from or is a predisposing factor to the development of ABPA. Patients with ABPA typically present with symptoms of asthma. Typical radiographic findings consist of central bronchiectasis with mucoid material filling the bronchi. The appearance of mucus-filled bronchi on chest radiography has been variously described as finger in glove, rabbit ears, toothpaste-shaped, and Y- or V-shaped opacities. The diagnosis of ABPA is based on major and minor criteria (35). Major criteria include asthma, blood eosinophilia, immediate skin reactivity to Aspergillus antigen, increased serum immunoglobulin E, transient or fixed pulmonary opacities on chest radiography, and central bronchiectasis. Minor criteria include fungal organisms in the sputum, history of expectoration of brown plugs or flecks, and delayed skin reactivity to fungal antigens. Because ABPA is an allergic disease, the primary treatment consists of corticosteroids.
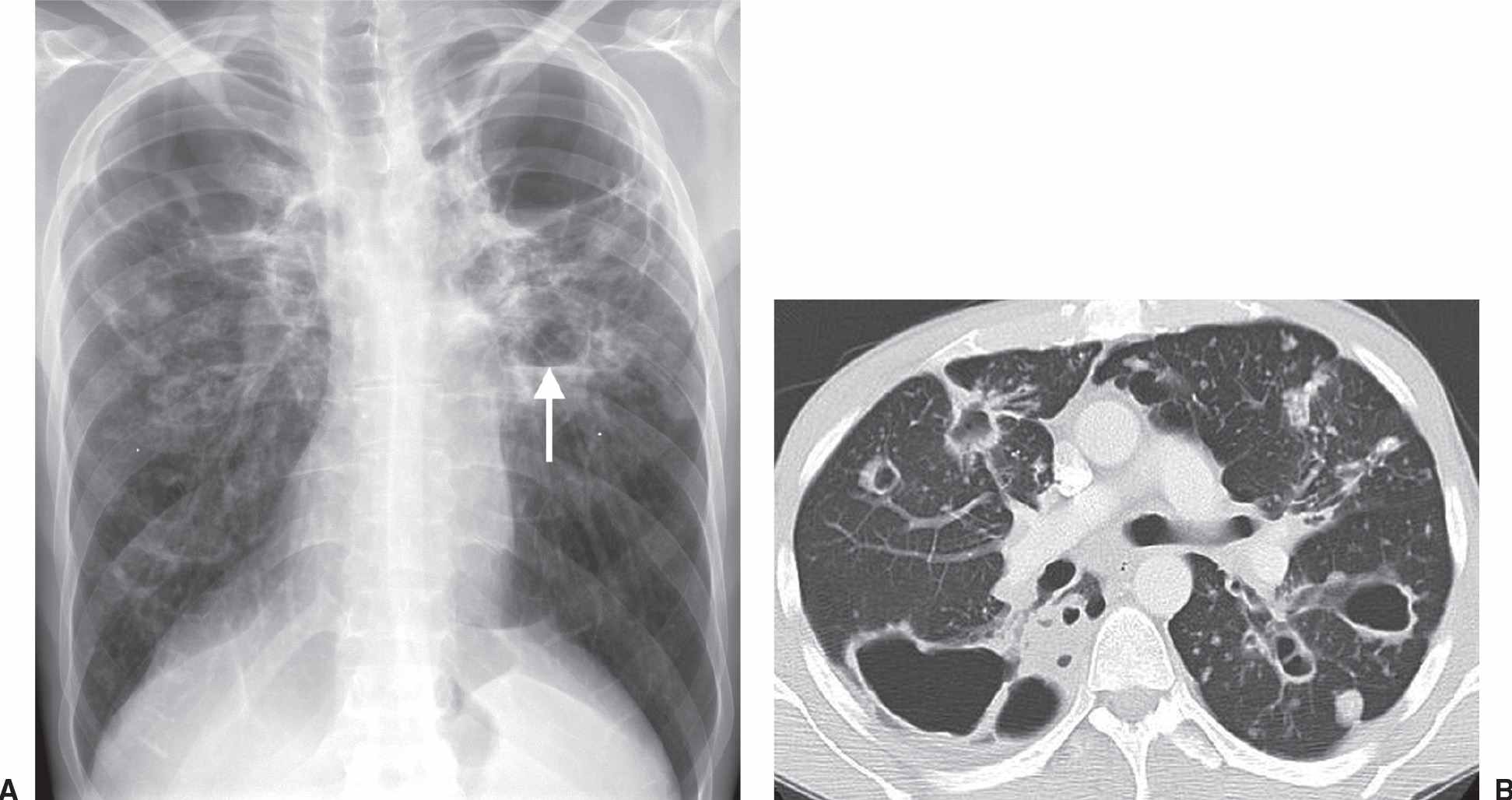
FIG. 10.30 • Mycobacterium avium infection. A: PA chest radiograph shows bilateral upper lung cavitary disease. An air–fluid level is present in the left upper lobe (arrow). B: CT shows multiple cavitary nodules of varying size.
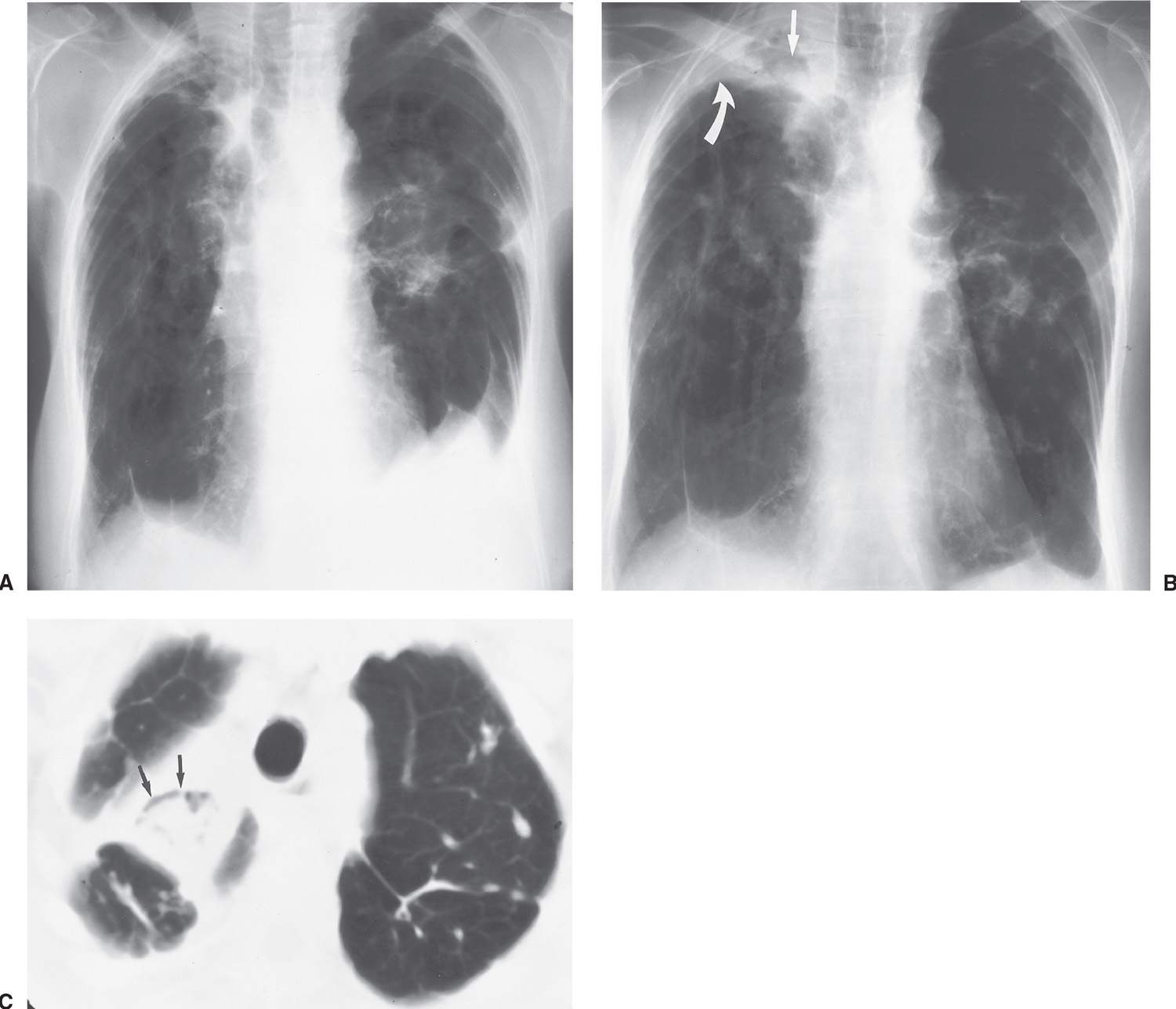
FIG. 10.31 • Noninvasive pulmonary aspergillosis. A: Baseline PA chest radiograph of a 79-year-old woman with class IV sarcoidosis shows retraction of the hila bilaterally, bilateral upper lobe fibrosis, and tenting of the hemidiaphragms from upper lobe volume loss. B: PA chest radiograph taken 2 years later shows increased right apical pleural thickening (curved arrow) and a fungus ball (aspergilloma) in a preexisting right upper lobe cavity (straight arrow). C: CT shows opacity within the right upper lobe cavity, representing vascular granulation tissue and fungal organisms. A small amount of air is seen within the cavity (arrows), creating the air crescent sign.
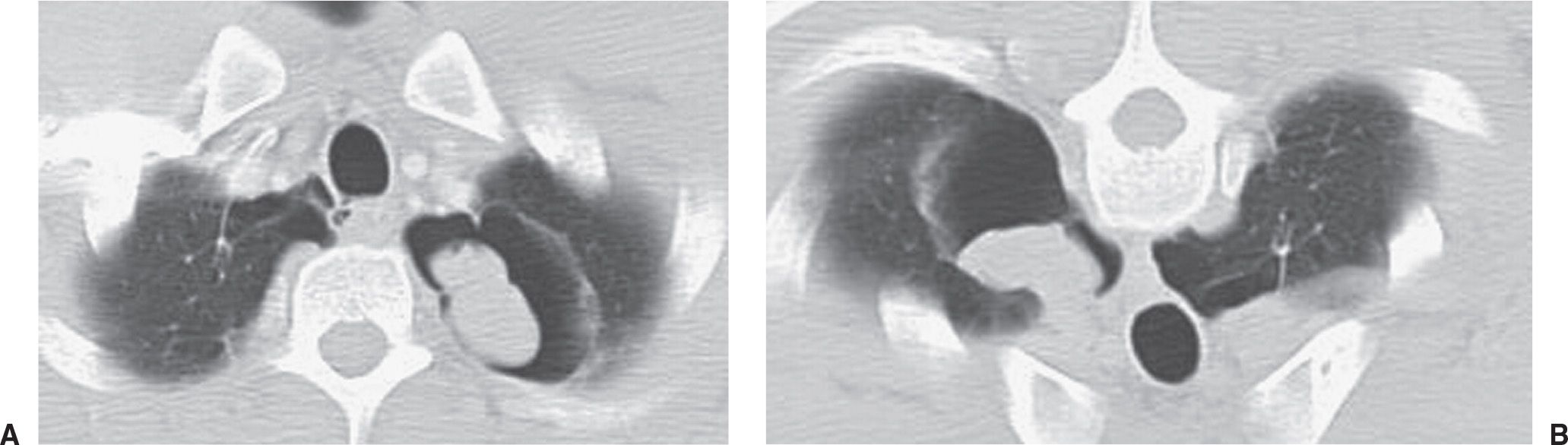
FIG. 10.32 • Noninvasive pulmonary aspergillosis. A: Supine CT image of a 41-year-old man with hemoptysis shows a thin-walled cavity in the left upper lobe containing an ovoid mass of soft tissue attenuation. B: Prone CT image shows movement of the mass to the dependent aspect of the cavity. Movement of an intracavitary fungus ball with change in patient positioning is typical of noninvasive aspergillosis.
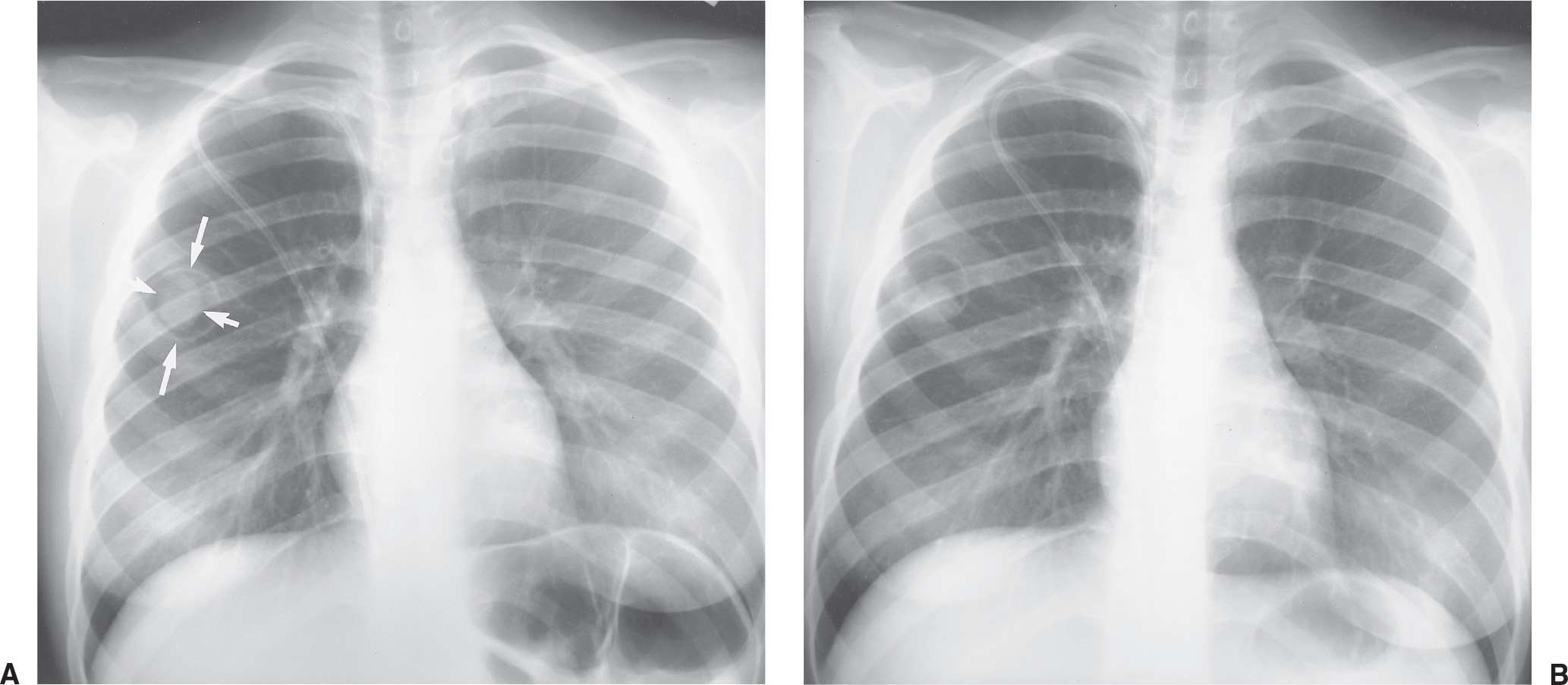
FIG. 10.33 • Noninvasive pulmonary aspergillosis. A: PA chest radiograph of a 14-year-old girl shows a thin-walled cavity in the right upper lobe (long arrows) containing a fungus ball (short arrows). B: PA chest radiograph after change in patient position shows movement of the mobile fungus ball.
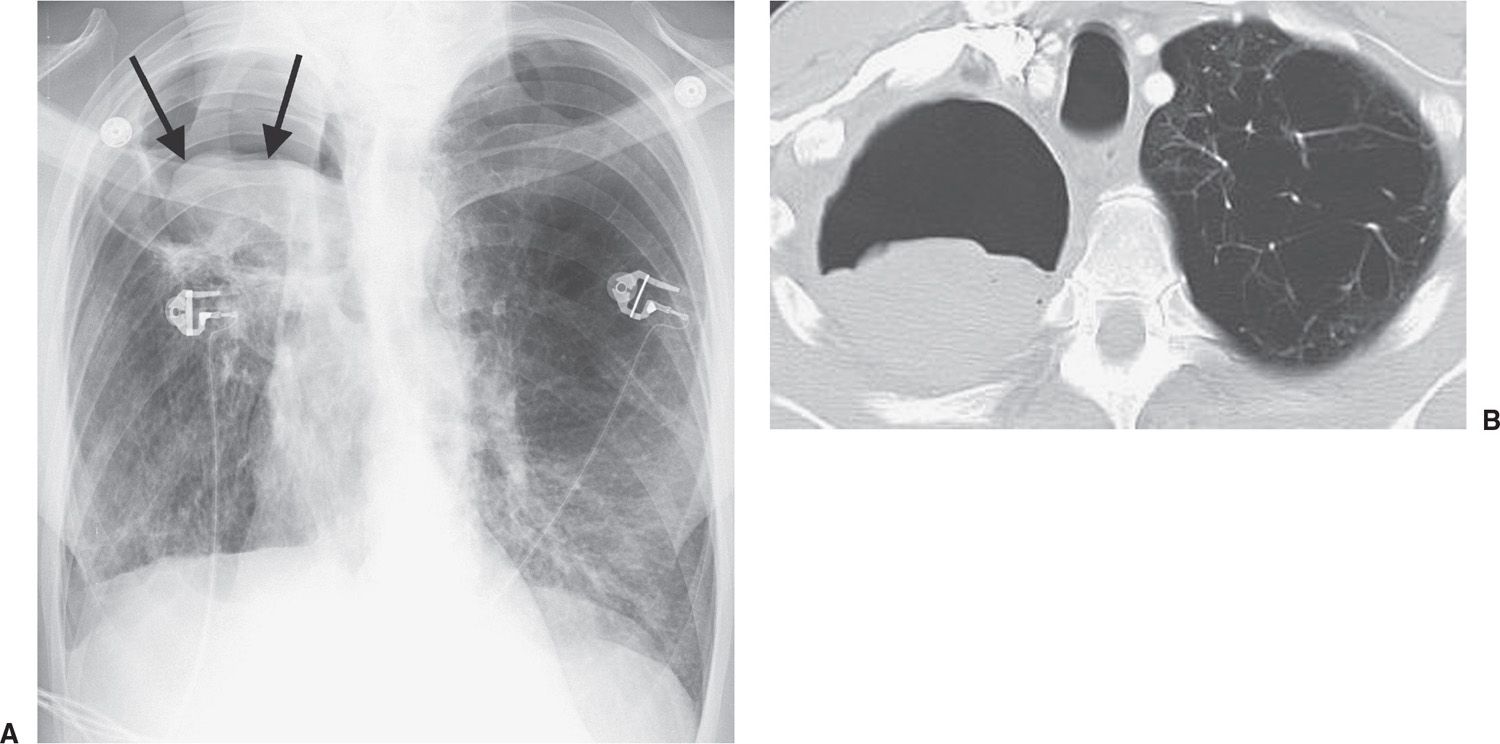
FIG. 10.34 • Semi-invasive pulmonary aspergillosis. A: PA chest radiograph of a 49-year-old woman with hemoptysis shows a rounded opacity (arrows) within a right upper lobe cavity. There is retraction of the right hilum along with elevation of the right hemidiaphragm. B: CT shows a fungus ball within the right upper lobe cavity. Note the severe changes of emphysema in the left lung.
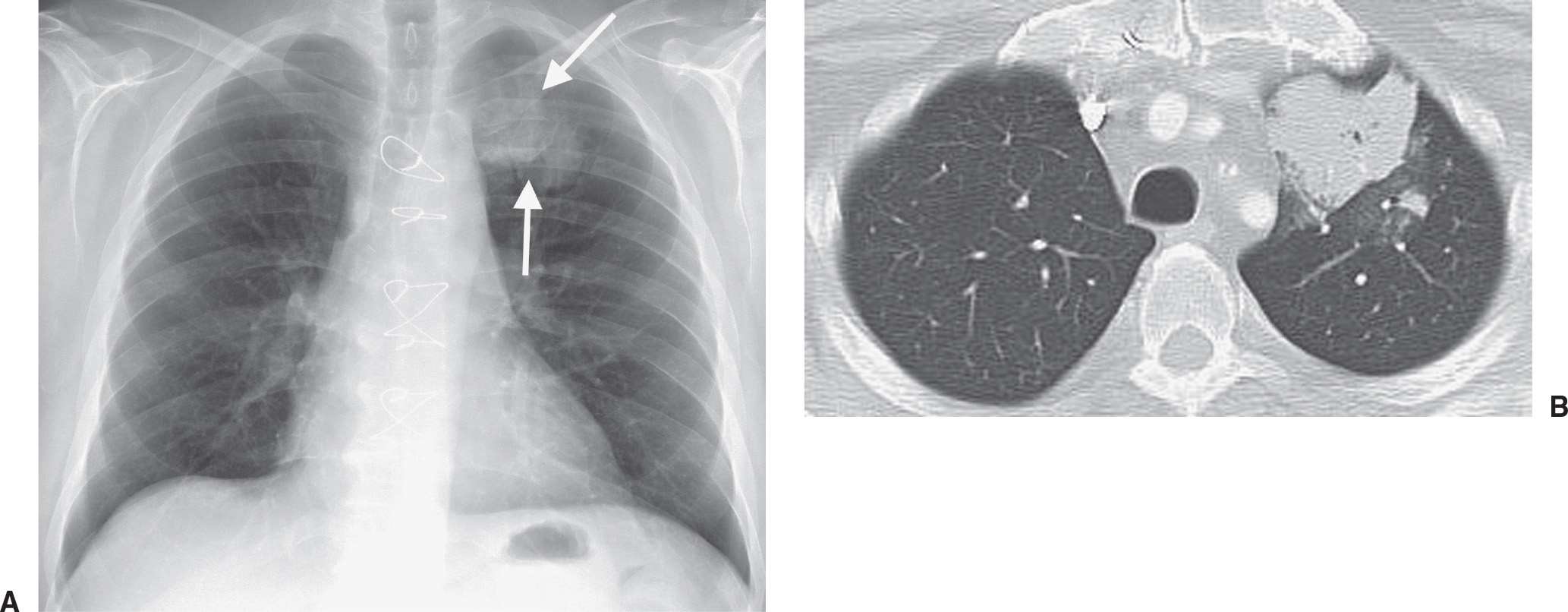
FIG. 10.35 • Invasive pulmonary aspergillosis. A: PA chest radiograph of a 57-year-old woman with a transplanted heart shows focal airspace opacity in the left upper lobe (arrows). B: CT shows dense airspace opacity and surrounding halo of ground-glass opacity in the left upper lobe.
Bronchocentric granulomatosis, characterized histologically by necrotizing granulomas that may obstruct and destroy bronchioles, is a histopathologic pattern that is generally believed to represent a nonspecific response to a variety of forms of airway injury. Approximately half of all cases are associated with asthma and ABPA, and among these patients, bronchocentric granulomatosis may represent a histopathologic manifestation of fungal hypersensitivity. Aspergillus hyphae can be identified within the granulomas in up to 50% of patients. Although the imaging features may be similar to those of conventional ABPA, the lung involvement in bronchocentric granulomatosis is more commonly focal and peripheral (36).
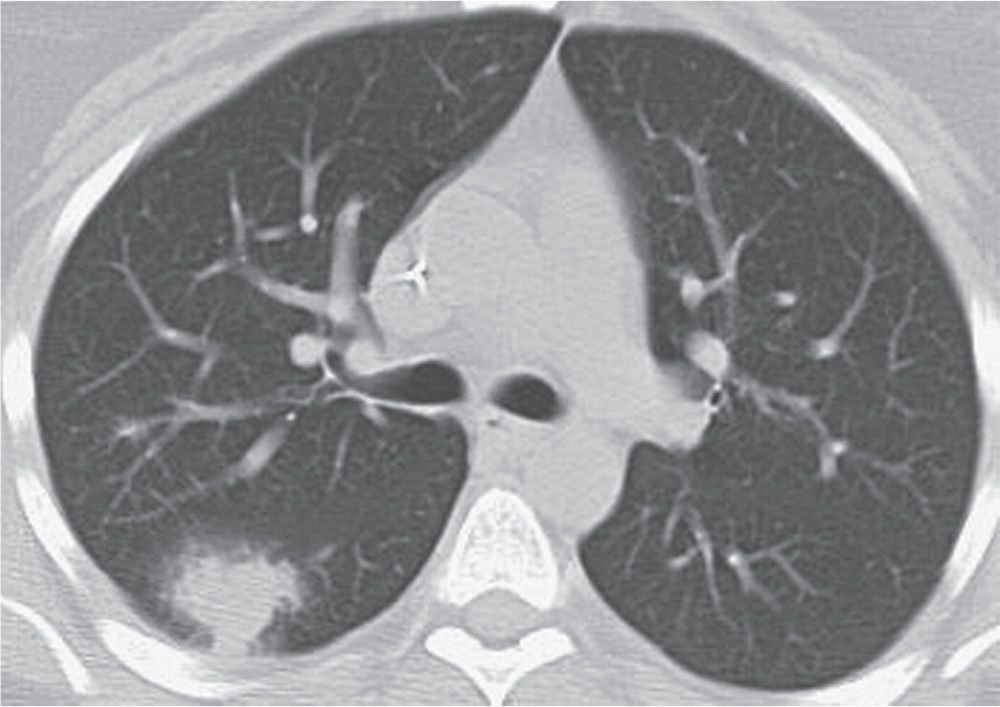
FIG. 10.36 • Invasive pulmonary aspergillosis. CT of a 30-year-old woman with leukemia and fever shows an ill-defined nodule with a halo of ground-glass opacity in the right lung.
CYSTIC FIBROSIS
Cystic fibrosis is an autosomal recessive disease involving chromosome 7 and characterized by dysfunction of exocrine glands, which form a thick, tenacious material. The incidence is 1 in 1,600 live births, with Caucasians affected most frequently. Organs involved include the lung, upper respiratory tract, pancreas, liver, gallbladder, and reproductive tract. The earliest chest radiographic findings include peribronchial inflammatory changes (peribronchial cuffing) and both atelectasis and hyperinflation. With progression of the disease, ring shadows and tram tracking are seen as signs of bronchiectasis, usually with an upper lobe predominance (Fig. 10.37). Not uncommonly, air–fluid levels can be seen in areas of cystic bronchiectasis. Small, ill-defined, or tubular opacities occur as a result of mucus plugging in dilated airways (Figs. 10.38 to 10.40). Larger opacities usually represent pneumonia, most commonly caused by Staphylococcus aureus or Pseudomonas sp. Bilateral hilar lymphadenopathy is common. Severe disease can lead to pulmonary arterial hypertension and cor pulmonale, which may be suggested when enlargement of the central pulmonary arteries and enlargement of the right heart, respectively, are seen on the chest radiograph. ABPA occurs in 10% of patients with cystic fibrosis, as discussed earlier in this chapter (Fig. 10.41). Hypertrophic pulmonary osteoarthropathy, a disorder involving the long bones and joints, occurs in approximately 15% of patients with cystic fibrosis.
RADIOLOGIC ABNORMALITIES IN IMMUNOCOMPROMISED PATIENTS
Infection is the leading cause of chest radiographic abnormalities in immunocompromised patients and is a direct result of a deficiency in immune defenses (e.g., immunoglobulin abnormalities, cell-mediated dysfunction, and phagocytic defense disorders) or a nonspecific reduction in host resistance (e.g., advanced age, alcoholism, diabetes, starvation or malnutrition, and cancer). In addition to infection, radiographic abnormalities in immunocompromised patients can result from many other causes, as listed in Table 10.4.
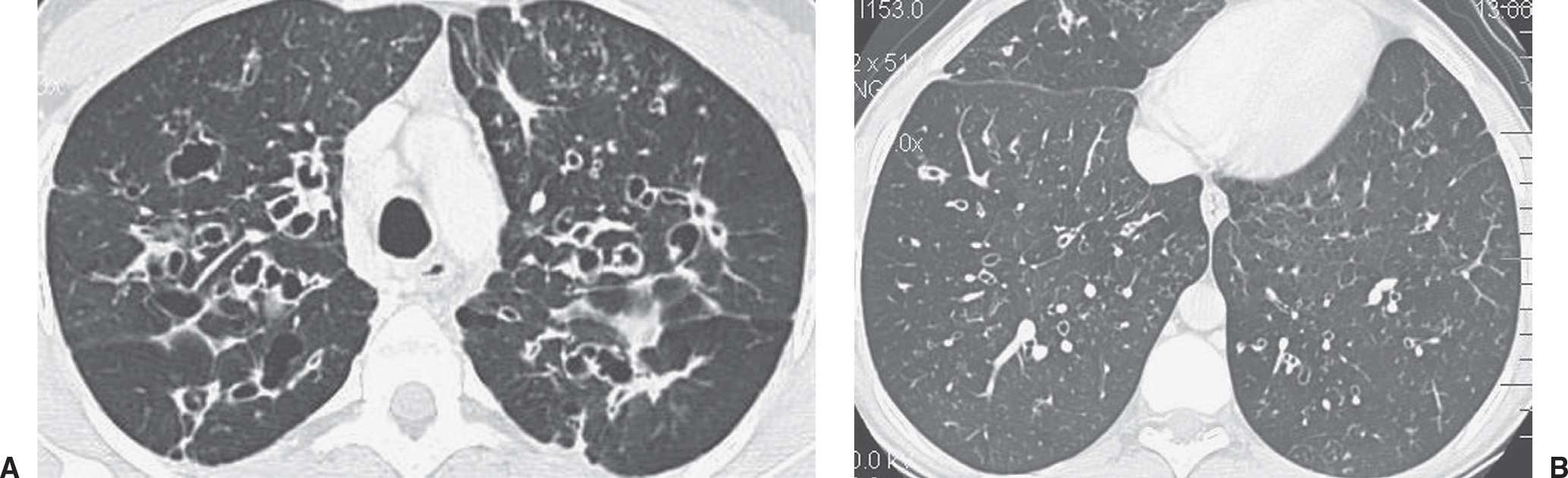
FIG. 10.37 • Cystic fibrosis. A: CT of a 29-year-old woman shows bilateral cystic bronchiectasis and thickening of bronchial walls. Small nodules in the left upper lobe likely represent mucoid-impacted bronchioles. B: CT at a more inferior level shows similar but less severe findings. Cystic fibrosis typically involves the upper lungs to a greater degree than the lower lungs.
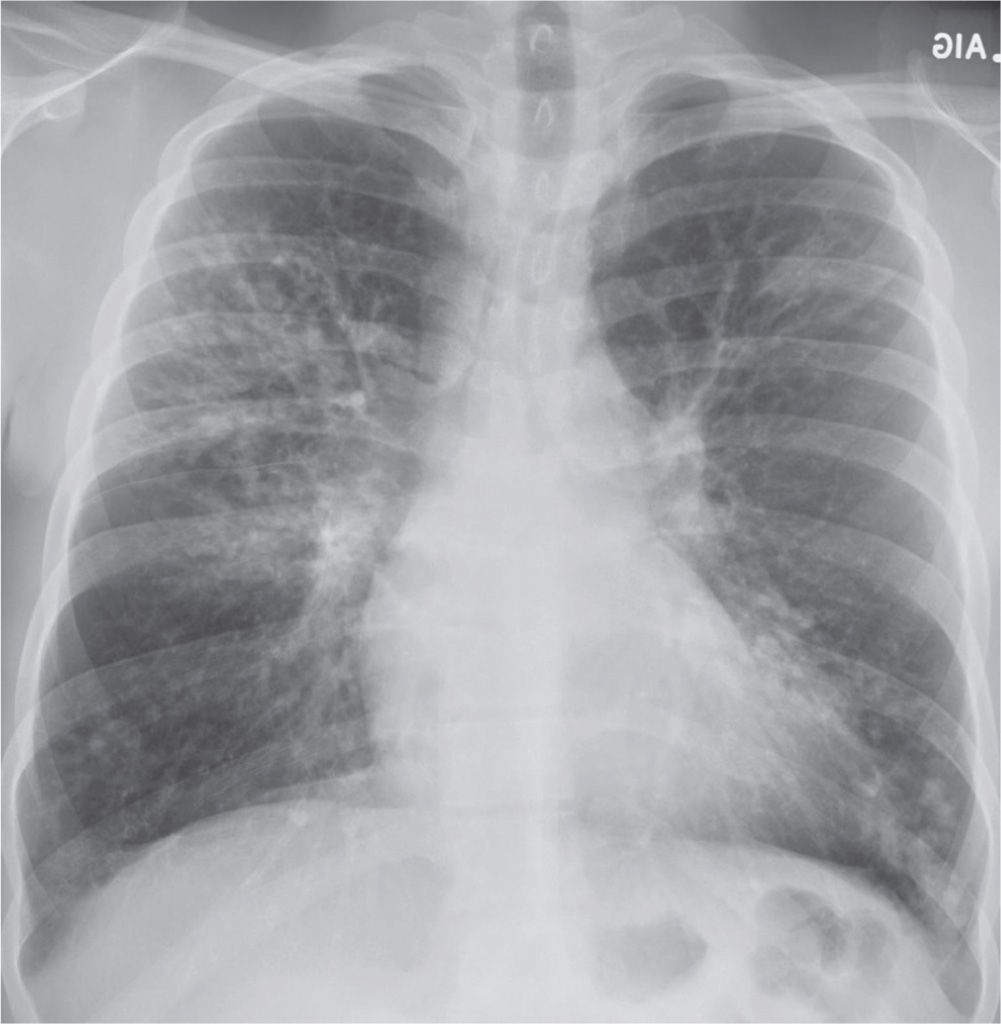
FIG. 10.38 • Cystic fibrosis. PA chest radiograph of a 22-year-old man with cystic fibrosis shows upper lung–predominant bronchiectasis, bronchial wall thickening, and scattered opacities representing mucoid impaction of airways.
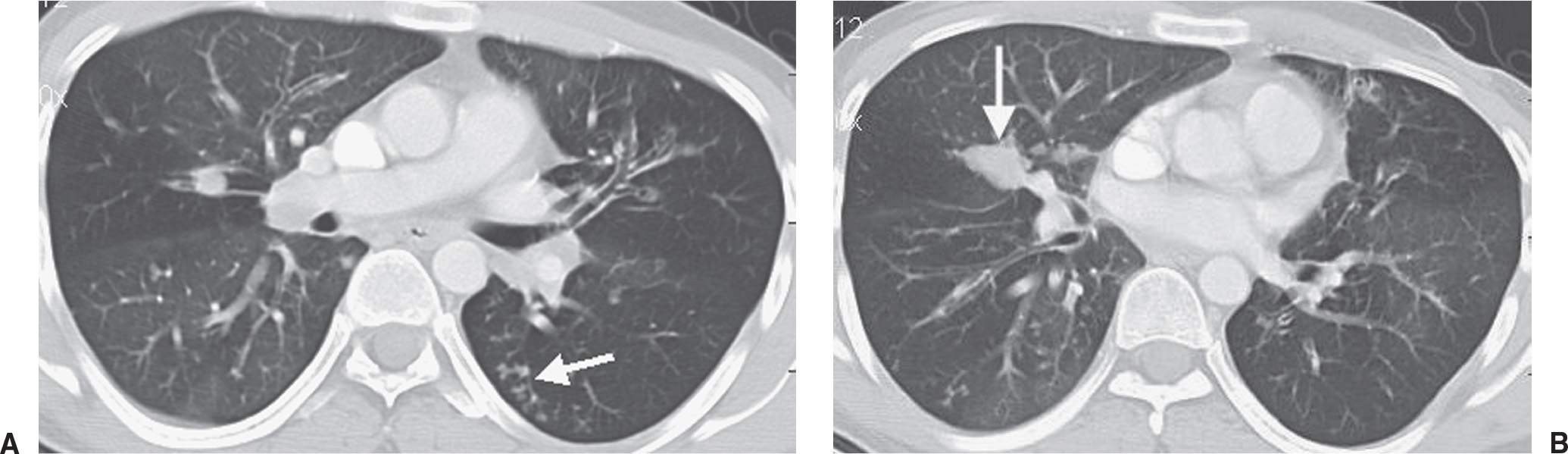
FIG. 10.39 • Cystic fibrosis. A: CT shows small nodular and linear branching opacities (arrow) in the left lower lobe, consistent with bronchiolar impaction. Note the bronchiectasis in both upper lobes. B: CT at a more inferior level shows a markedly distended, mucoid-impacted bronchus in the right middle lobe (arrow). This finding is suggestive of ABPA, which patients with cystic fibrosis are predisposed to develop.
Infection
Bacterial agents are the most frequent causes of pneumonia in immunocompromised patients. Patients receiving steroids and patients with renal transplants are particularly susceptible to infection with Legionella organisms. Legionella pneumophila manifests as progressive parenchymal consolidation, sometimes with cavitation and pleural effusion. Legionella micdadei pneumonia results in well-circumscribed nodular densities with central cavitation (37). Nocardia asteroides infection occurs most commonly in immunosuppressed patients, including those with AIDS (Fig. 10.42) and solid organ transplants. Chest radiographs and CT scans show lobar or multilobar areas of consolidation, or they show solitary or multiple ill-defined pulmonary nodules with a tendency to cavitate and invade surrounding structures such as the chest wall (Fig. 10.43) (38,39).
TB in immunocompromised patients occurs most commonly in the AIDS population. In non-AIDS immunocompromised patients, TB is usually caused by reactivation of a dormant lesion. The radiographic features typically consist of apical and posterior segmental fibronodular disease in the upper lobes with or without cavitation (40). Infection with atypical mycobacteria can have a similar appearance, but there is a tendency toward increased cavitation relative to total lung involvement, thin-walled cavities with less dense surrounding parenchymal opacification, less bronchogenic and more contiguous spread, more involvement of the apical and anterior segments of the upper lobes, and marked pleural thickening over the involved areas of the lung (40).
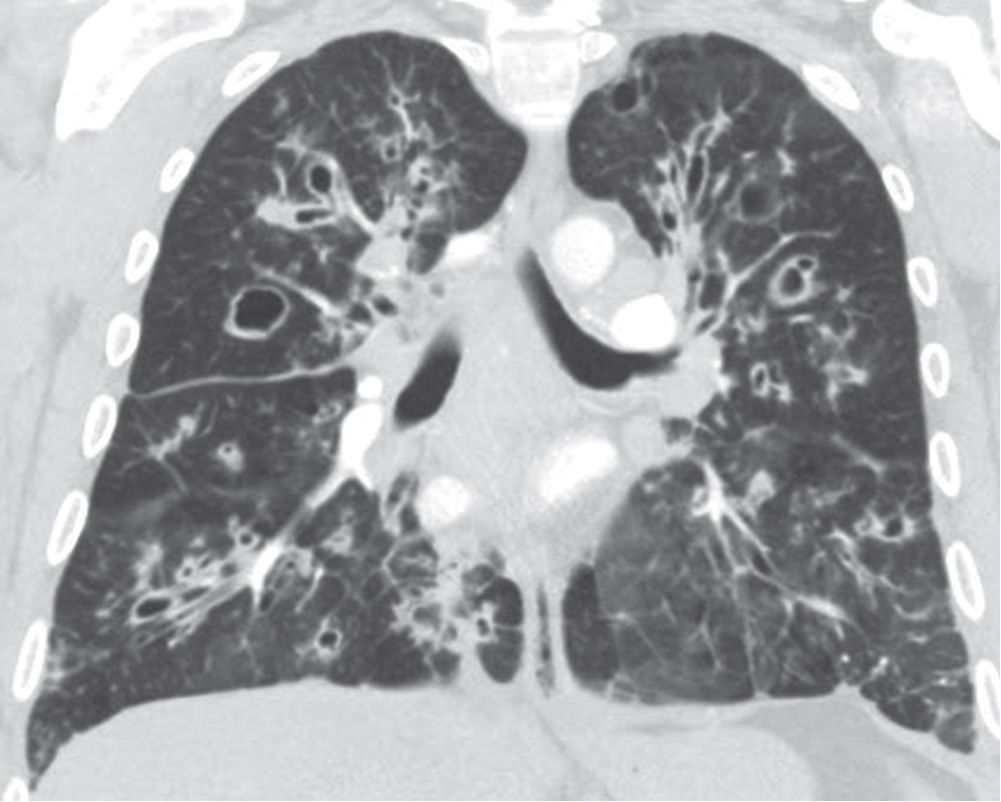
FIG. 10.40 • Cystic fibrosis. Coronal CT shows bilateral bronchiectasis, bronchial wall thickening, and areas of mucoid impaction.
Aspergillus fumigatus is an important fungal pathogen in immunocompromised patients, especially those with lymphoma or leukemia. The different types of Aspergillus lung disease were discussed earlier in this chapter.
Candida albicans is another fungal agent that causes pneumonia in a substantial number of patients with leukemia or lymphoma. Chest radiographs most commonly show diffuse, bilateral, nonsegmental, patchy alveolar or mixed alveolar–interstitial opacities. A pattern of miliary opacities can also be seen (41).
Mucormycosis results from infection with one of the phycomycetes and has a 100% mortality rate in the absence of treatment. Patients susceptible to mucormycosis include those with leukemia or lymphoma and those with diabetes mellitus. Pathologically, mucormycosis is fungal vascular invasion resulting in infarction (42). The most common radiographic abnormality is a single pulmonary nodule, mass, or focus of consolidation, frequently with cavitation. This is usually accompanied by infection of the paranasal sinuses, brain, and meninges.
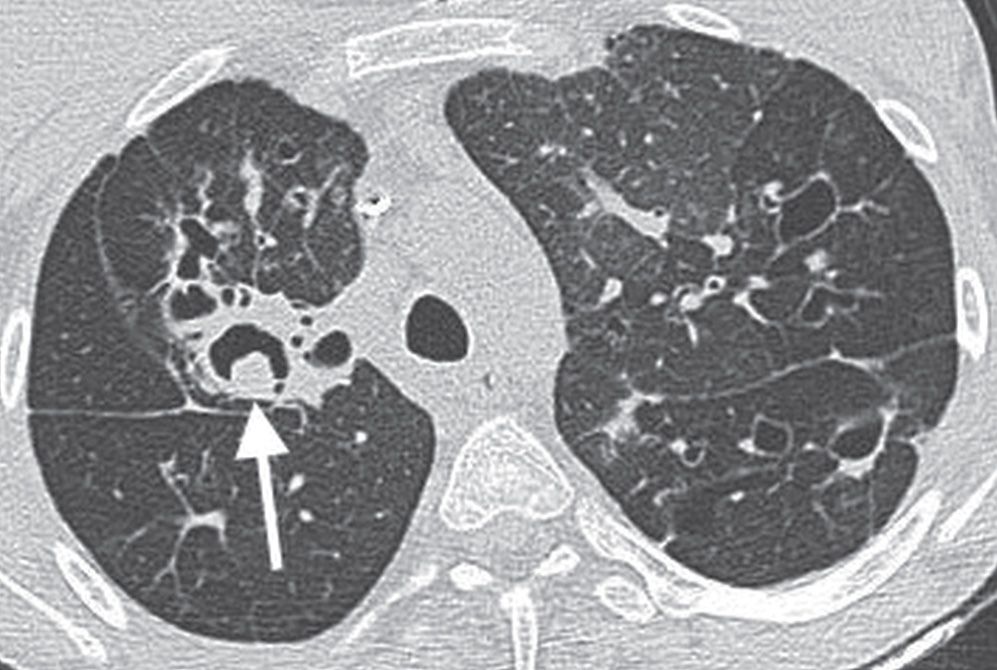
FIG. 10.41 • Cystic fibrosis and ABPA. CT of a 21-year-old woman with cystic fibrosis shows a dilated airway containing Aspergillus hyphae in the right upper lobe (arrow), consistent with allergic bronchopulmonary aspergillosis (ABPA).
Table 10.4 CAUSES OF RADIOGRAPHIC ABNORMALITIES IN IMMUNOCOMPROMISED PATIENTS
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


