CHAPTER 22 Venous Occlusive Disease
The term cerebral venous occlusive disease signifies partial to complete occlusion of one or more of the dural venous sinuses, deep cerebral veins, or superficial cerebral veins on an acute, subacute, or chronic basis. Other names include cerebral venous thrombosis (CVT), dural sinus thrombosis (DST), deep cerebral venous thrombosis (DCVT), and superficial cerebral venous thrombosis (SCVT).
EPIDEMIOLOGY
The annual incidence of cerebral venous occlusive disease may be as low as two to seven cases per million in the general population.1,2 Autopsy series estimate the prevalence of CVT from less than 1% to as high as 9% of patients dying of stroke.3 CVT tends to affect women more than men and has a younger age distribution than arterial thrombosis, with a particularly high incidence in neonates. Most patients have underlying congenital or acquired hypercoagulable states, but the disease may result from local vascular compression or be idiopathic. The prognosis for recovery is better than that for arterial infarction.
CLINICAL PRESENTATION
The signs and symptoms of CVT are highly variable, depending on the acuity of thrombosis, its location and extent, and the availability of collateral pathways. Clot propagation and spontaneous thrombolysis occur simultaneously in as many as 70% of patients,4 complicating the clinical picture. Overall, CVT presents acutely in approximately 30% of cases (<2 days from onset of symptoms), subacutely in approximately 50% of patients (from 2 to 30 days), and chronically in 20% of patients (>30 days).5 The acute presentation may be abrupt or rapidly progressive. Because of this variation, venous thrombosis may mimic the temporal course of acute cerebral infarction or subarachnoid hemorrhage (abrupt onset), acute encephalitis (rapidly progressive over 1 to 3 days), brain abscess (progressive abnormalities developing over 1 to 2 weeks), or brain tumor and neurodegenerative disease (slow progression over months to years). Therefore, CVT is often underdiagnosed or misdiagnosed.
The symptoms of CVT can be nonfocal or focal. Nonfocal findings typically result from DST, secondary venous hypertension, and raised intracranial pressure. Headache is reported by 74% to 90% of patients and is typically the first manifestation of CVT. The headache can develop gradually or suddenly and can mimic the typical “thunderclap” presentation of subarachnoid hemorrhage.6 Other nonfocal findings include papilledema (45%), seizures with possible Todd paresis (29%), confusion and coma (26%), as well as nausea, vomiting, visual changes, and cranial nerve palsies.2,3 Overall, 20% to 40% of patients with CVT present with signs and symptoms of intracranial hypertension, so the possibility of CVT should be considered in any patient with raised intracranial pressure.2 Idiopathic intracranial hypertension must be considered only as a diagnosis of exclusion.
In most cases, the thrombus is formed within the sinus and subsequently extends retrograde into adjacent deep or cortical veins. Isolated thrombosis within a deep or superficial vein without sinus thrombosis is uncommon. In DST, focal deficits such as hemiplegia, dysphasia, paresthesias, and cranial nerve palsies may occur in conjunction with the nonfocal symptoms of intracranial hypertension. Focal or generalized seizures, followed by hemiparesis, aphasia, hemianopia, or other focal deficits occur without the signs of intracranial hypertension typically seen with DST.7 In cavernous sinus thrombosis, presenting symptoms include headache, periorbital edema, proptosis, chemosis, and abnormalities of eye movements secondary to compromise of nerves that run through the cavernous sinus (oculomotor, trochlear, and abducens). Patients with isolated SCVT present with focal neurologic deficits dependent on the location of the occlusion. Patients with DCVT often present with altered consciousness and signs referable to the corticospinal tract and other long white-matter tracts. They may have a more rapidly declining clinical course with poor outcomes more common, including death and long-term morbidity.8 Symptoms secondary to severe dysfunction of the diencephalon, such as coma and abnormalities of eye movements and reflexes, are more common.8 In one small series, five of seven patients with DCVT presented with headache, nausea, vomiting, and altered level of consciousness.9 However, clinical findings and outcome in DCVT are variable. On occasion, presentation can be relatively mild with headache and no disturbance of consciousness. If deep venous thrombosis is rapidly reversed (by spontaneous or therapeutic clot lysis), patients may have complete recovery.
In contrast to arterial ischemic stroke, relief of venous obstruction, even if delayed, has been shown to yield dramatic clinical improvement in particular with regard to signs and symptoms of increased intracranial pressure. The median time from the onset of symptoms to the correct diagnosis of CVT is reported to be as much as 7 days.10 Shortening the time from symptom onset to diagnosis has important prognostic implications because early diagnosis and treatment (anticoagulation and/or endovascular thrombolysis) can lead to recovery with surprisingly few sequelae. Resolution of edema in patients with CVT with normalization of the apparent diffusion coefficient has been reported after treatment.11
PATHOPHYSIOLOGY
There are many causes of CVT (Table 22-1). These are subclassified into local processes such as infection, which hinder local blood flow, and systemic processes such as pregnancy and the puerperium, oral contraceptives, coagulopathies, and inflammatory bowel disease, which lead to hypercoagulable states.12 Approximately 25% of patients with CVT have detectable genetic bases for their hypercoagulable state. Inherited hypercoagulability should be suspected if a patient has recurrent CVT, is younger than 45 years of age, has a family history of venous thrombosis, or has no acquired risk factors. The most common genetic risk factor for CVT is factor V Leiden, followed by the prothrombin gene mutation G20210A. Other less common inherited causes of CVT are deficiencies of protein S, protein C, and antithrombin III.13 In as many as 33% of patients, no cause can be identified.12
TABLE 22-1 Etiology of Cerebral Venous Thrombosis
CVT does not occur in an even distribution throughout the cerebral venous system. In a large study of 624 patients the distribution frequency of CVT was superior sagittal sinus, 62%; left and right transverse sinuses, 44.7% and 41.2%, respectively; straight sinus, 18%; isolated cortical veins, 17.1%; deep venous system, 10.9%; cavernous sinus, 1.3%; and cerebellar veins, 0.3%.10 Multifocal thromboses, particularly in the contiguous transverse and sigmoid sinuses, are present in as many as 33% of patients.2 Thirty to 40 percent of patients with DST have concurrent thrombosis of adjacent cerebral veins.14
Thrombus typically begins within the dural sinuses with subsequent retrograde extension into the cortical veins that drain into that sinus. A four-step process has been described in a pig model of superior sagittal sinus thrombosis. Nonocclusive thrombus forms within the dural sinus, leading to eventual occlusion. Subsequently, there is extension of thrombus into the cortical veins. It is hypothesized that the extension into cortical veins is the key event leading to pathologic changes in the brain parenchyma secondary to lack of sufficient collateral drainage.15,16 The pathophysiology described in this model likely applies to humans as well. In many cases, isolated sinus thrombosis is tolerated until secondary involvement of cortical and medullary veins causes more substantial impairment of cerebral venous drainage and more significant clinical signs and symptoms. This model may also explain why the extent of the sinus thrombus does not always correlate with the extent of the resulting lesion, because extension into feeding veins is more important in determining the severity of the parenchymal lesion.17
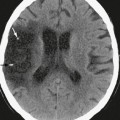
Local effects secondary to occlusion of cerebral veins include focal edema, hemorrhage, and venous infarction. The acute phase of occlusion is marked by venous engorgement and prominence of collateral veins. If venous drainage cannot be adequately supplied by these collateral vessels, venous pressure rises, leading to breakdown of the blood-brain barrier and leakage of blood serum into the interstitial space (vasogenic edema). As pressures continue to increase, tearing of thin-walled medullary veins results in hemorrhage. Hemorrhage occurs most commonly in the parenchymal subcortical white matter near the thrombosed sinus. Hematoma formation leads to mass effect on adjacent brain parenchyma and rupture of neural pathways. Less commonly, hemorrhage occurs in the subarachnoid and subdural spaces, presumably due to rupture of cortical and bridging veins as they course through these spaces. This pathophysiology has been validated by studies correlating pressure measurements within a thrombosed sinus and the presence of edema and hemorrhage. Hemorrhage was present with intrasinus pressures higher than 42 mm Hg, whereas lower pressures produced edema without hemorrhage.18 Increased venous pressure may also lead to decreased arterial regional cerebral blood flow (rCBF) that, when severe or prolonged, produces ischemia with resultant ion-pump failure, cytotoxic edema, and cell death (venous infarction). MRI has demonstrated restricted diffusion in brain lesions resulting from CVT, and it has been estimated that venous infarction occurs in about 50% of patients with CVT.10
After thrombus formation and sinus occlusion, thrombus breakdown can lead to spontaneous partial or complete recanalization. This recanalization process tends to occur earlier rather than later, with one study at 3 weeks after CVT demonstrating 40% complete recanalization and 20% partial recanalization.19 After 4 months of anticoagulation therapy, there is typically no further resolution.19 Recanalization after anticoagulation therapy occurs more often with thrombosis of the superior sagittal and straight sinuses as opposed to thrombosis of the transverse and sigmoid sinuses.19 Complete recanalization is not necessary for clinical recovery, and the extent of recanalization may not correlate closely with the clinical outcome.19 This is likely secondary to the numerous venous collateral pathways that form after sinus thrombosis, which typically include dural collaterals that surround the occluded sinus, cortical venous collaterals that drain into other patent sinuses, and diploic, emissary, and meningeal collaterals as well.2
An interesting yet poorly understood aspect of the pathophysiology of chronic CVT is related to the formation of dural arteriovenous fistulas (AVFs). Arterial growth into the wall of the occluded vein leads to recanalization of chronic CVT with formation of AVFs. Dural AVFs may produce hemorrhage or infarction if venous pressure is high and cortical veins are dilated. Chronic venous hypertension may present as cognitive decline that mimics neurodegenerative disease. This is a rare but well-established long-term complication of CVT and should be sought in patients with CVT who undergo subsequent imaging studies.20
PATHOLOGY
Acute thrombus in the superior sagittal sinus is identical to other thrombi, with fresh clot rich in red blood cells and fibrin. The fresh clot is eventually replaced with fibrous tissue. The vessel may recanalize. The affected brain shows subcortical hemorrhage, infarction, and edema. Histologic analysis reveals hemorrhage or hemorrhagic infarction. Typically, venous infarctions show more pronounced leukocyte infiltrations than do arterial infarctions, because the patent artery allows inflow of white cells.21 In protracted cases with chronic VST, venous hypertension causes marked fibrous thickening and hyalinization of the walls of the veins draining into the sinus.22
The venous anatomy of the brain is variable and complex. This variability underlies the wide range of clinical and imaging features of CVT. In considering this anatomy, one can divide the system into three components: (1) superficial venous drainage, (2) deep venous drainage, and (3) dural venous sinuses (Figs. 22-1 to 22-3).
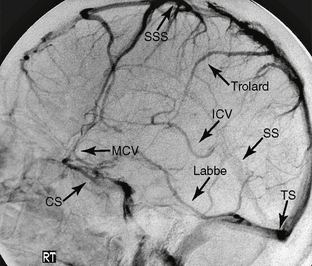
FIGURE 22-1 Normal venous anatomy as seen on lateral projection DSA. CS, Cavernous sinus; SSS, superior sagittal sinus; MCV, middle cerebral vein; ICV, internal cerebral vein; TS, transverse sinus.
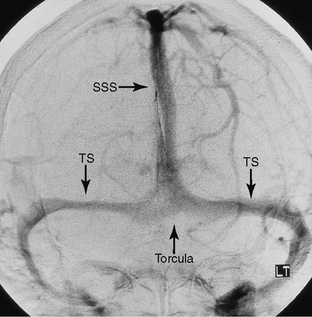
FIGURE 22-2 Normal venous anatomy as seen on anteroposterior projection DSA. SSS, superior sagittal sinus; TS, transverse sinus.
Superficial Drainage
The superficial cerebral veins drain the cerebral cortex and the adjacent white matter into the dural venous sinuses. The superficial veins are subclassified by their location and by the dural sinus into which they most often drain: (1) superior group draining predominantly into the superior sagittal sinus; (2) sphenoidal group draining predominantly into the sphenoparietal then cavernous sinuses; (3) tentorial group draining predominantly into the transverse sinus, and (4) the falcine veins, which drain predominantly to the deep and midline drainage system.23
The falcine group is composed of veins that drain into the deep venous system, including the internal cerebral veins, the basal veins of Rosenthal, and the great vein of Galen. Cortical areas drained by the falcine group correspond to the limbic system and include the parahippocampal, cingulate, paraterminal, parolfactory gyri, and the uncus. The venous drainage from the region of the sylvian fissure deserves special mention. Drainage occurs via the superficial sylvian vein into the sphenoparietal sinus along the sphenoid ridge, as well as the veins of Trolard and Labbé. Because these veins communicate with each other, their relative caliber is inversely proportional.23
Deep Drainage
The deep venous system consists of the internal cerebral, vein, the basal vein of Rosenthal, and the vein of Galen and their respective tributaries. The deep system drains the deep white and gray matter around the ventricles and structures around the basal cisterns. It can be divided into a ventricular group that drains the walls of the ventricles and a cisternal group that drains the structures around the cisterns.23 In forming the ventricular group, subependymal and thalamostriate veins course along the walls of the ventricles and converge on the internal cerebral, basal, and great veins. Veins draining the frontal horn and the body of the lateral ventricle generally drain into the internal cerebral vein, whereas veins originating from the temporal horn generally drain into the basal vein. Veins from the ventricular atrium drain into all three veins. The cisternal group of deep veins drains the area beginning just anterior to the third ventricle and extending laterally and posteriorly to the sylvian fissure and quadrigeminal plate cistern. The main structures drained by the deep system include the inferior frontal lobe; most of the deep white matter of the frontal, temporal, and parietal lobes; the corpus callosum; the upper brain stem; the basal ganglia; the choroid plexus; the hippocampus; the cortical areas of the limbic lobe (cingulate and parahippocampal gyri); the visual cortex; parts of the cerebellum; and the thalamus. Knowledge of these drainage patterns is important in accurately diagnosing deep venous obstruction. As a direct result of this anatomy, deep venous obstruction spares the subcortical white matter of the cerebral convexities; cortical involvement is limited to the inferior frontal, limbic lobe, and visual cortex.23,24
The basal vein of Rosenthal is an important component of the deep venous system. It arises near the uncus by the confluence of the anterior and deep middle cerebral veins. Although the basal vein drains into the vein of Galen, it communicates with the superior petrosal sinus (via the lateral mesencephalic vein), with the cavernous sinus and pterygoid plexus in adults (via the deep and superficial sylvian veins), and on occasion with the straight or transverse sinus. Communications between the basal vein and the vein of Galen through choroidal, thalamic, and striate anastomoses can divert outflow from the deep system in the case of vein of Galen occlusion. Because of these anastomotic interconnections, only simultaneous obstruction of the vein of Galen and the basal vein of Rosenthal effectively obstructs deep venous outflow.8 This situation most commonly arises near their confluence, in the posterior aspect of the tentorial incisura. Any lesion that causes sufficient mass effect on the midbrain may cause such venous obstruction. Complete obstruction of the great vein of Galen and basal veins leads to rapid death. Incomplete obstruction leads to various degrees of injury within areas drained by these deep veins.25