Fig. 1
Scatterplot showing the relationship between tumor size and pimonidazole-positive fraction (PPF) for HT29 tumors. The x-axis represents effective tumor diameter (D) derived from the cross-sectional area (A) of H&E images, D = 2√A/π (Li et al. Cancer Res 2007;67:7646)
Micrometastases may have already existed in many patients when primary cancers were initially diagnosed although lack of imaging evidence of distant metastases. Submillimeter metastases in patients may be avascular and in a state of dormancy [27, 28]. Cell proliferation in dormant tumors had been observed, but results were mixed: cells were either found dividing very slowly or in G0 phase [27–30], others have found proliferation in dormant tumors to be as high as in macroscopic vascularized tumors but that the dormant tumors did not grow beyond a threshold size due to a kinetic balance between proliferation and apoptosis [28, 31], and hypoxia status had not been observed in dormant micrometastases in these studies. In animal model of metastases, we have noted that cellular proliferation was in the nonhypoxic rim, not the interior hypoxic core of submillimeter avascular tumors, whereas proliferating cells were found throughout larger tumors 1–4 mm in diameter which were less hypoxic [8, 9, 25]. This is in good agreement with several studies that cellular proliferation and hypoxia are mutually exclusive in macroscopic tumors [32–34]. Future studies would confirm whether cancer cells in dormant metastases are proliferative; this issue is critically important for systemic chemotherapy, as chemotherapeutic drugs generally target proliferating cancer cells.
Hypoxia has been recognized as a primary physiological regulator of angiogenesis [35, 36]. The existence of severe hypoxia in microscopic tumors may be common irrespective of cell lines and reflects the pre-angiogenic stage of tumor development. As peritoneal and intradermal tumors increased in size to the diameter range ~1–4 mm, there was a drastic reduction in tumor hypoxia coupled with the appearance of functional tumor vascularization [8, 9, 25]. This suggests the sequence of events may be that cells become hypoxic when tumors reach several hundred micrometers, hypoxia drives angiogenesis, previously hypoxic cells become oxygenated, and the neovascularized tumors grow beyond certain size threshold. Apparently, the timing for hypoxia driving angiogenesis switch is critically important for anti-angiogenesis therapy. This is largely unknown in metastases involving other organs and tissues rather than peritoneal cavity.
Animal Models of Metastases
To conduct preclinical study of tumor microenvironment, animal models of cancer and metastases are required. Models of disseminated microscopic malignant disease may be generated in the lung, bone, liver, and peritoneum by intravenous [37–39], left ventricular [37, 40], intrasplenic [41], and intraperitoneal [8, 9, 11, 12, 25] injection of tumor cells, respectively (Fig. 2). We have shown that a model of disseminated microscopic peritoneal and ascites tumors was suitable for studying hypoxia [8, 9, 11, 12, 25]. In the model, disseminated peritoneal microscopic tumors were induced by injecting suspensions of colorectal HT29 and HCT-8 cancer cells or NSCLC A549 and HTB177 cells into the peritoneal cavity of nude mice. Mice sacrificed 4–7 weeks after tumor initiation displayed a distribution of tumors of sizes ranging from a few hundred micrometers up to several millimeters in diameter on or in the intestinal serosa. Ascites fluid containing a distribution of free-floating tumor cell aggregates of up to 1 mm in diameter was frequently presented in mice inoculated with HT29, A549 cells, but was rarely observed in HCT-8 and HTB177 cell lines. Intradermal injection of similar tumor cell suspensions could give rise to unitary microscopic tumors useful for studying hypoxia [8, 25]. An intradermal tumor model has the potential advantage that growth curves for microscopic tumors could be more easily generated via ultrasound or optical imaging. Subcutaneous xenografts generated by subcutaneously injection of cancer cells are widely used for PET study.
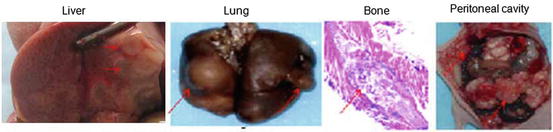
Fig. 2
Nude mice metastatic models
Imaging Hypoxia in Metastases
We have recently reviewed that a model system is valid for assessing the microenvironmental features of individual microscopic tumors with targeted PET tracers, including hypoxic status, proliferation, and blood perfusion [10]. Such model system provides a diversity of tumors of differing size and hypoxic status growing in the same animal, thereby reducing or eliminating issues associated with inter-animal variability. To complement the high spatial resolution associated with immunohistochemical detection of hypoxia markers, the intratumoral distribution of radiolabeled hypoxia tracers can be determined by digital autoradiography. Studies comparing the spatial distributions of radiolabeled tracers and hypoxia markers have been reported in macroscopic tumors [22, 33] and microscopic diseases [8].
18F-misonidazole [17–19] and 124I-IAZGP [23, 24, 42] among others are used for in vivo PET imaging of tumor hypoxia. We have recently reported the use of correlative imaging methodologies to examine the uptake of IAZGP and 18F-misonidazole in microscopic [10, 12] and macroscopic [11, 12] tumors and related this to microenviromental factors. Pimonidazole binding and CA9 expression in a small (~1 mm dimension) and relatively large tumor obtained from an animal with disseminated colorectal cancer HT29 peritoneal disease, the smaller tumor shows elevated uptake of 131I-IAZGP and near-ubiquitous staining of pimonidazole and CA9, implying severely hypoxia. The larger tumor shows reduced 131I-IAZGP uptake and low levels of pimonidazole binding and CA9 expression with significant Hoechst 33342 uptake indicating that the tumor was well perfused. Microscopic ascites tumors had high 131I-IAZGP uptake coupled with intense staining of pimonidazole and CA9 [10].
Intratumoral distribution of 18F-misonidazole was observed in subcutaneous xenografts and peritoneal tumors of NSCLC; subcutaneous xenografts and peritoneal metastases were generated utilizing human NSCLC A549 and HTB177 cell types in nude mice. High levels of 18F-FMISO uptake detected by digital autoradiography and pimonidazole binding by immunohistochemical staining were colocalized [11]. Such regions tended to correspond to low levels of cellular proliferation and blood perfusion. Well-oxygenated cancer cells with a high proliferation rate had low 18F-misonidazole uptake. Stroma and necrotic zones had lower 18F-misonidazole accumulation [11, 12].
In a PET study [12], we compared the intraperitoneal accumulation of 18F-misonidazole between peritoneal disease-free mice and mice with A549 or HTB177 peritoneal metastases. All image sets for each animal were visually examined using a rotating (cine) three-dimensional display. Figure 3 shows representative PET coronal slices: In metastases-bearing mice, high radioactivity accumulation was found on the left side of abdominal wall, and the overall background in the peritoneal cavity (excluding the intestines) was apparently higher than in the normal mouse. Necropsy revealed that the left peritoneal wall was the site of multiple individual tiny lesions. In both the disease-free and metastases-bearing mice, there was significant uptake of 18F-misonidazole in the gut and bladder and to a lesser extent in the liver. Tumors from the left peritoneal wall were removed for sectioning, H&E staining demonstrated multiple individual micrometastases (generally less 1 mm in diameter), and some of them fused together. The micrometastases had little Hoechst 33342 uptake, indicating a lack of blood perfusion, and a high fraction of pimonidazole binding and high 18F-misonidazole accumulation, indicating severely hypoxic tissue. There was little 18F-misonidazole accumulation in the stroma. Ascites was collected from the peritoneal cavity, and single cells or ascites tumors were harvested by centrifugation, and these stained positive for pimonidazole. Therefore, 18F-misonidazole PET is able to detect micrometastases in the peritoneal cavity [12].
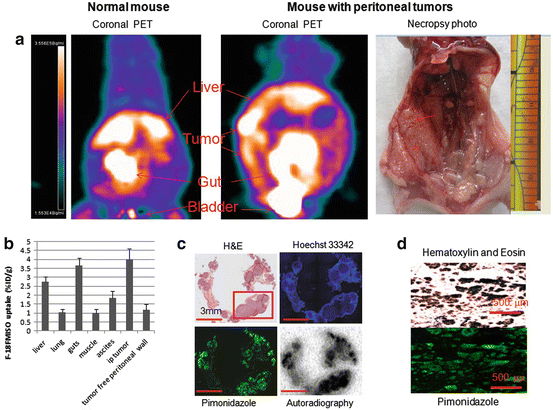
Fig. 3
(a) PET coronal slices of intraperitoneal distribution of 18F-misonidazole in a peritoneal disease-free mouse (left) and a mouse with peritoneal metastases after A549 cell inoculation (middle) with necropsy- confirmed peritoneal wall carcinomas (right). High radioactivity accumulation was found on the abdominal wall as indicated, and the overall background in the peritoneal cavity (excluding guts) was apparently higher than control. (b) 18F-misonidazole (18F-FMISO) distribution in A549 i.p. tumors in mice (n = 5). (c) Multiple individual lesions attaching to the peritoneal wall where PET imaging had revealed a high level of 18F-misonidazole: Hematoxylin and Eosin stain showing multiple individual micrometastases (generally less 1 mm in diameter) which had little Hoechst 33342 uptake, associated with a high fraction of pimonidazole binding and high 18F-misonidazole accumulation by autoradiography. (d) Single cells or ascites tumors harvested from ascites of the mouse were stained positive for pimonidazole, Hematoxylin and Eosin stain provides as a reference. All scale bars are as indicated. Adapted from [12]
Figure 4 shows a representative 18F-misonidazole PET mid-coronal slice of a macroscopic A549 subcutaneous tumor, showing considerable heterogeneity in the spatial distribution of the tracer. Autoradiography, pimonidazole, glucose transporter-1 expression, and Hoechst 33342 images were obtained on the same section, and H&E staining was performed on an adjacent section. 18F-misonidazole colocalized with pimonidazole, which was roughly similar to glucose transporter 1, and these regions mutually excluded Hoechst 33342. Regions of low pimonidazole binding also had low 18F-misonidazole uptake. Necrosis and stroma were also associated with low 18F-misonidazole activity. Therefore, 18F-misonidazole accumulated in hypoxic cancer cells, and low radioactivity regions were either nonhypoxic cancer tissue or stroma and necrosis [12].
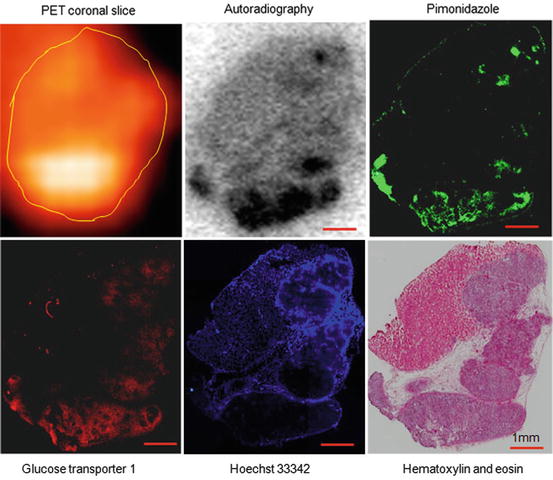
Fig. 4
Intratumoral distribution of 18F-misonidazole in a macroscopic A549 subcutaneous xenograft by PET and autoradiography and its relationship to tumor microenvironment. Autoradiography, pimonidazole, glucose transporter-1 expression, Hoechst 33342, and Hematoxylin and Eosin stains were obtained from the same frozen tissue section. Stroma and pimonidazole-negative cancer cells associated with low 18F-misonidazole accumulation. Glucose transporter-1-expressing regions are much wider than those positive for pimonidazole. All scale bars are 1 mm. Adapted from [12]
In summary, 131I-IAZGP and 18F-misonidazole have been validated as hypoxia imaging tracers in microscopic and macroscopic tumor models colorectal cancer and NSCLC.
Imaging Glucose Metabolism with 18F-FDG
18F-FDG PET has emerged as an important clinical tool for cancer detection, staging, and monitoring of response and is routinely used in the clinical management of several cancer types. The uptake of 18F-FDG, an analog of glucose, is largely proportional to the rate of glucose metabolism enabling this parameter to be quantified [43]. In hypoxic conditions, cancer cells may undergo a switch from aerobic to anaerobic glucose metabolism. This adaptive response involves the coordinated expression of many HIF-regulated proteins, such as glucose transporters 1, and various glycolytic enzymes [44].
In vitro experiments show that incubation in hypoxic conditions induces an increase in cellular 18F-FDG uptake [45–47]. It was recently shown that the intratumoral distribution of 18F-FDG in R3327-AT rat prostatic carcinoma xenografts positively correlated with that of the hypoxic marker pimonidazole [33]. We have recently observed glucose uptake in microscopic tumors grown intraperitoneally in nude mice using 18F-FDG digital autoradiography and to relate this to physiological hypoxia and glucose transporter-1 expression [25]. Human colon cancer HT29 and HCT-8 cells were injected intraperitoneally into nude mice to generate disseminated tumors of varying sizes. Following overnight fasting, animals, either breathing air or carbogen (a gas mixture of 95 % O2 and 5 % CO2), were intravenously administered 18F-FDG together with the hypoxia marker pimonidazole and the cellular proliferation marker bromodeoxyuridine 1 h before sacrifice. Hoechst 33342, a perfusion marker, was administered 1 min before sacrifice. The intratumoral distribution of 18F-FDG was assessed by digital autoradiography of frozen tissue sections. This was compared with the distributions of pimonidazole, glucose transporter-1 expression, bromodeoxyuridine, and Hoechst 33342 as visualized by immunofluorescent microscopy. In air-breathing condition, small tumors (generally less than 1 mm in diameter) had high 18F-FDG accumulation and were severely hypoxic with high glucose transporter-1 expression and low proliferation. Larger tumors (1–4 mm diameter) generally had low 18F-FDG accumulation and were not significantly hypoxic with low glucose transporter-1 expression but high proliferation. Carbogen breathing significantly decreased 18F-FDG accumulation and tumor hypoxia in microscopic tumors but had little effect on glucose transporter-1 expression. We concluded that micrometastases have high 18F-FDG uptake, therefore, high glucose demand which is spatially associated with physiological hypoxia and high glucose transporter-1 expression. This enhanced uptake was abrogated by carbogen breathing, indicating that in the absence of physiological hypoxia, high glucose transporter-1 expression, by itself, was insufficient to ensure high 18F-FDG (glucose) uptake [10, 25].
Therefore, 18F-FDG uptake was significantly increased in microscopic peritoneal tumors and only hypoxic regions of macroscopic tumors [11]. This enhanced uptake could be abrogated by carbogen breathing; physiological hypoxia was a necessary condition for increased 18F-FDG uptake.
Intratumoral distribution of 18F-FDG and 18F-misonidazole detected by autoradiography was similar and that both tracers accumulated in hypoxic zones [11]. We extended our observation of 18F-FDG and 18F-misonidazole using PET on the same tumor-bearing animals. 18F-FDG and 18F-misonidazole PET scans were performed separated by a 24 h interval in the same animals. The intratumoral distribution of 18F-FDG and 18F-misonidazole was roughly similar, although areas of mismatch were apparent [12].
Imaging Proliferation with18F-Fluorothymidine
Related posts:
 Approach to Detection and Characterization of Hepatic Hemangiomas
Approach to Detection and Characterization of Hepatic Hemangiomas
 Resonance Imaging of Adenocarcinoma of the Pancreas
Resonance Imaging of Adenocarcinoma of the Pancreas
 Imaging Characteristics of Hepatocellular Carcinoma
Imaging Characteristics of Hepatocellular Carcinoma
 Cortex Segmentation on Computed Tomography
Cortex Segmentation on Computed Tomography
 Liver Segmentation Without Prior Statistical Models
Liver Segmentation Without Prior Statistical Models
 Surface Registration Using Ricci Flow
Surface Registration Using Ricci Flow
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


