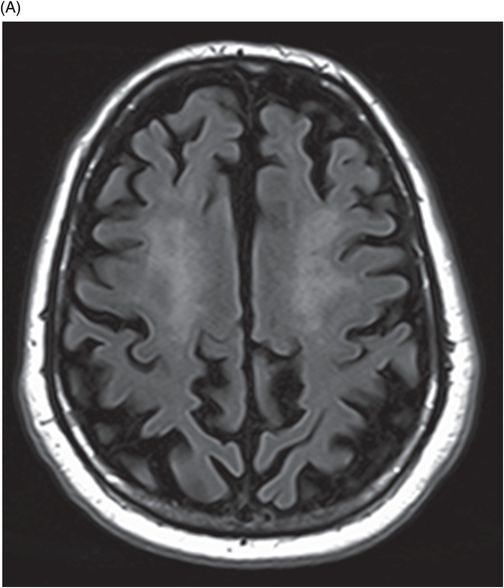
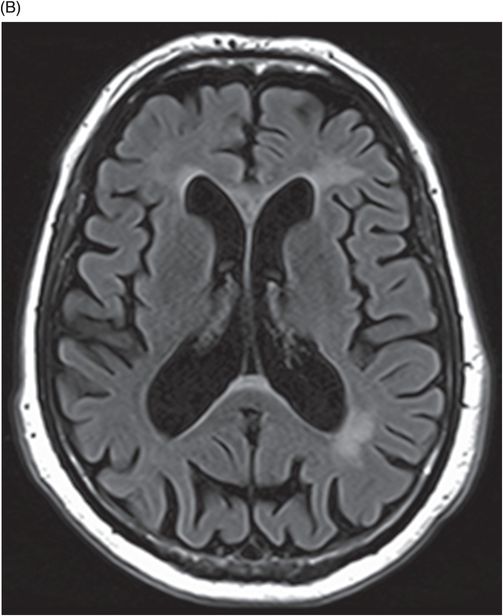
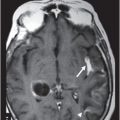
(A–B) Axial FLAIR images at the level of corona radiata, centrum semiovale, and lateral ventricles.
Fragile X Tremor Ataxia Syndrome
Primary Diagnosis
Fragile X tremor ataxia syndrome
Differential Diagnoses
Multiple system atrophy
Dentatorubral-pallidoluysian atrophy
Wilson disease
Progressive multifocal leukoencephalopathy (PML)
Post-ischemic changes
Imaging Features
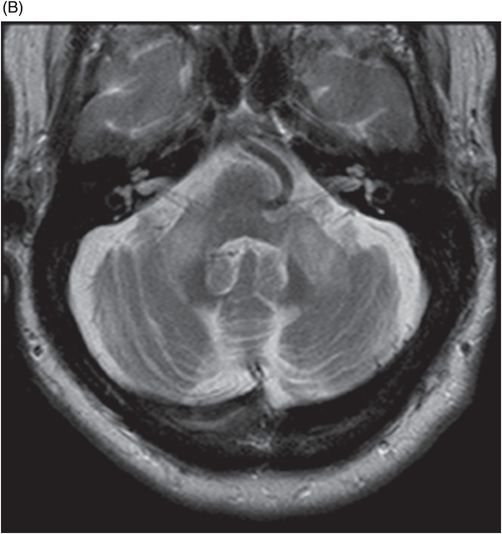
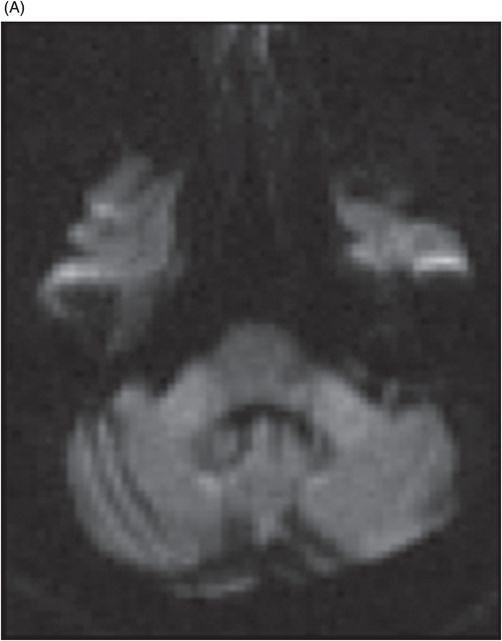
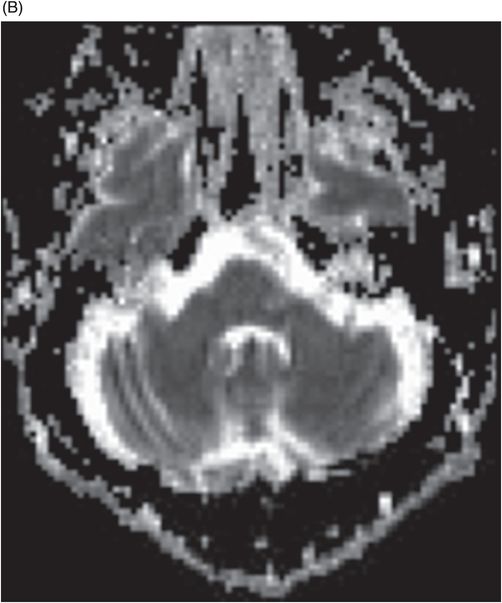
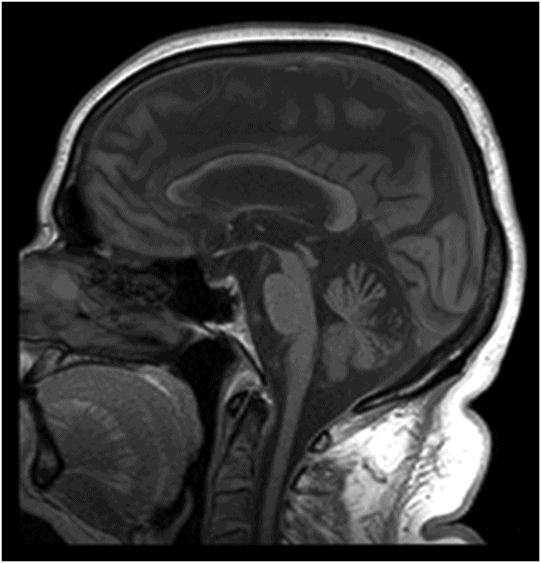
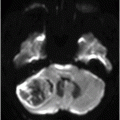
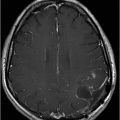
Fig. 122.1: (A) Axial FLAIR and (B) Axial T2-weighted images showed nearly symmetric hyperintense signal changes in the bilateral middle cerebellar peduncles. Generalized cerebellar volume loss was also noted. Fig. 122.2: (A) Axial DWI and (B) Corresponding ADC map did not show diffusion restriction. Fig. 122.3: (A–B) Axial FLAIR images at the level of the corona radiata, centrum semiovale, and lateral ventricles demonstrated generalized volume loss with deep white matter changes. Fig. 122.4: Midsagittal T1WI demonstrated thinning of the corpus callosum and cerebellar volume loss. The overall morphology and volume of the brainstem, especially the pons, is relatively preserved.
Discussion
Progressive intentional tremors and gait ataxia (non-responsive to anti-Parkinsonism medications), cognitive decline, autonomic disturbances, and nearly symmetric T2W FLAIR hyperintense signal changes in the middle cerebellar peduncles, with diffuse cerebral and cerebellar volume loss on neuroimaging, are the key clinicoradiologic features suggestive of fragile X tremor ataxia syndrome (FXTAS).
The list of differentials for symmetric middle cerebellar peduncle hyperintense signal changes is extensive. Multiple system atrophy exhibits similar findings; however, FXTAS demonstrates less severe pontine volume loss and can be confirmed by genetic testing. Dentatorubral-pallidoluysian atrophy (DRPLA), a CAG trinucleotide repeat neurodegenerative disorder, manifests with dementia, seizures, and choreoathetosis. The age of DRPLA onset is variable and based on the juvenile, early adult, or late-onset types. On imaging it has cerebral, cerebellar, and brainstem volume loss, with white matter changes. The central pons often shows signal changes on T2W and FLAIR sequences with sparing of the middle cerebellar peduncles. The age of onset is more than 50 years in FXTAS and, unlike DRPLA, is associated with CGG trinucleotide repeat sequences. Wilson disease can have similar clinical features; however, the age of presentation, lack of key imaging findings involving the basal ganglia and midbrain, and presence of normal ceruloplasmin levels are some of the salient findings that negate Wilson disease as a diagnosis in this case. Middle cerebellar peduncle hyperintensities can be noted in infectious demyelinating pathologies such as PML; however, negative HIV status and CSF analysis exclude these pathologies. Bilateral middle cerebellar peduncle infarcts are a rare but well-known entity and have similar T2W and FLAIR signal changes; however, the lack of diffusion restriction and duration of clinical presentation do not favor a vascular pathology.
FXTAS is a late-onset X-linked dominant neurodegenerative disorder with variable penetrance. It occurs as a result of a mutation in the FMR1 gene (fragile X mental retardation) that affects the CGG trinucleotide repeat sequence and ultimately causes a deficiency of the FMR1 protein (FMRP). FXTAS, commonly seen in males between 50 and 80 years of age, leads to mental retardation and autism. Although it primarily affects males, females with premutation carriers can manifest with features of ovarian failure in 20% of cases. In the general population, the estimated carrier rate of FMR1 premutation is approximately 1/259 for females and 1/800 for males. Literature describes the overall penetrance, which increases with age, in males to be around 40% at 50 years of age. However, the degree of penetrance in females is estimated at approximately 10%.
The clinical presentation includes intentional tremor, ataxia, neuropathy, and personality changes in association with memory loss. A combination of clinical presentation, genetic testing, and neuroimaging findings has been put forth as the criteria for diagnosis of FXTAS (see Tables 122.1, 122.2).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


