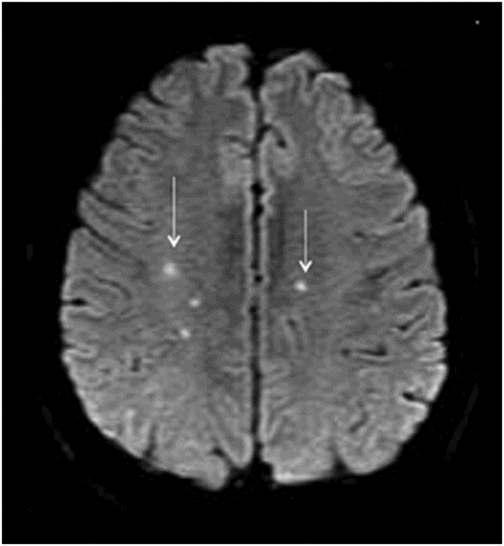
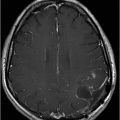
Axial T2WI at the level of centrum semiovale/corona radiata.
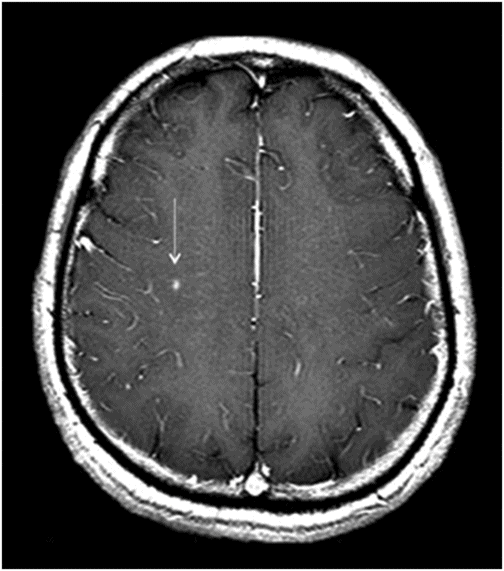
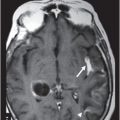
Axial postcontrast T1WI at the level of centrum semiovale/corona radiata.
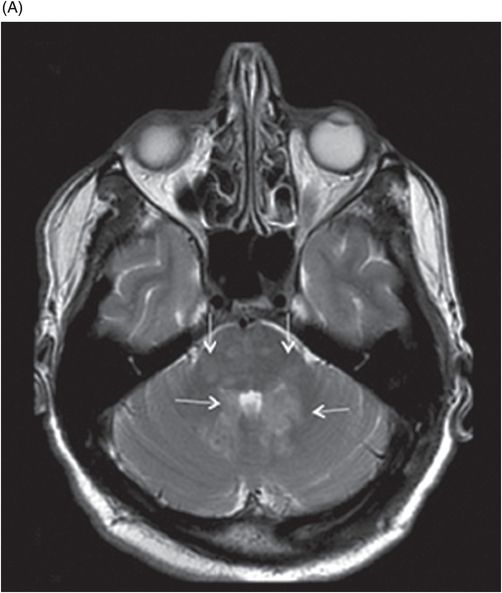
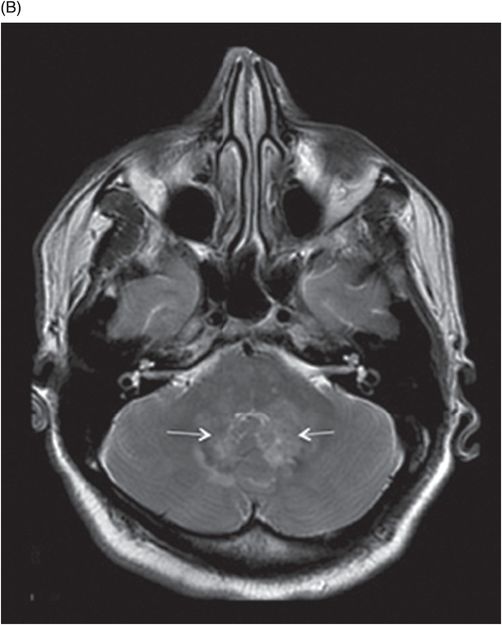
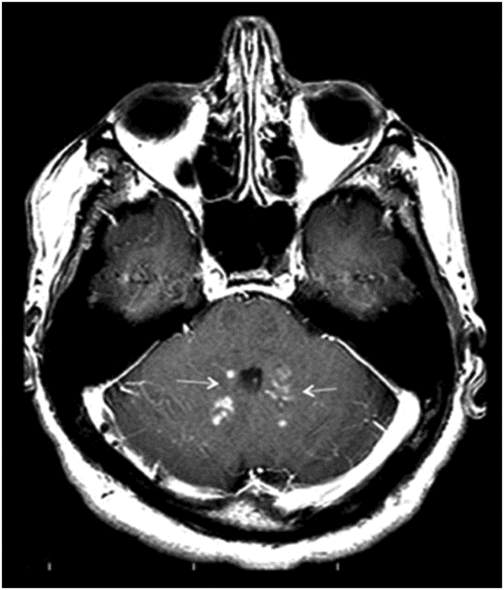
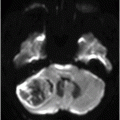
(A–B) Axial T2W images at the level of pons, cerebellar hemispheres, middle cerebellar peduncles, and dentate nuclei.
Erdheim-Chester Disease
Primary Diagnosis
Erdheim-Chester disease
Differential Diagnoses
Acquired and inherited causes of adult-onset ataxia
Glioma
Multiple sclerosis
Lymphoma
Sarcoidosis
Imaging Findings
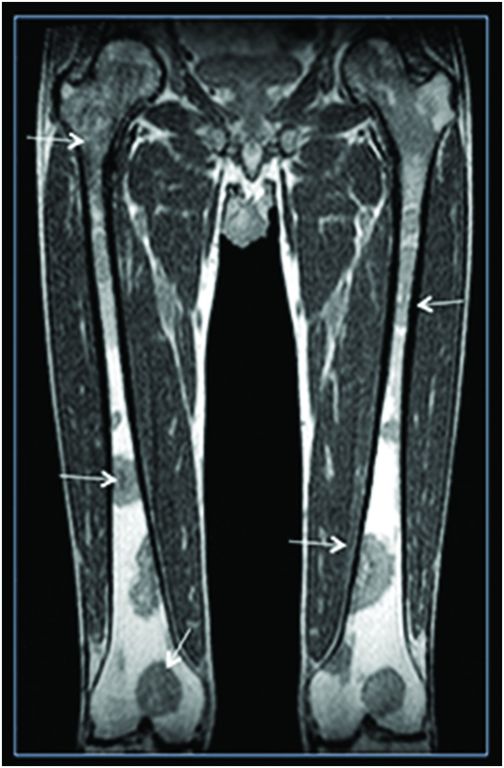
Fig. 131.1: T2W hyperintense lesions were noted in the central deep white matter (arrows). Fig. 131.2: Few of these lesions show diffusion restriction on DWI sequence. Fig. 131.3: Postcontrast T1WI shows nodular enhancement (arrow) in the right centrum semiovale. Fig. 131.4: (A–B) Similar confluent T2-hyperintense lesions are seen in the pons, bilateral superior and middle cerebellar peduncles, and dentate nuclei (arrows). Fig. 131.5: Nodular enhancement was seen in the bilateral cerebellar dentate nuclei (arrows). Fig. 131.6: Coronal T1WI demonstrates geographic pattern hypo- to isointense lesions predominantly involving the diametaphyseal regions of bilateral femora (arrows). Biopsies of bone and skin tissue were positive for lymphocytes, macrophages, and lipid-laden histiocytes, thus confirming the diagnosis of ECD.
Discussion
The differential diagnoses of Erdheim-Chester disease (ECD)-related progressive cerebellar syndrome include alcoholic cerebellar degeneration, paraneoplastic cerebellar degeneration, immune-mediated ataxias, acquired vitamin deficiency, superficial siderosis, chronic CNS infections, spinocerebellar ataxia, fragile X-associated tremor/ataxia syndrome (FXTAS), and multiple system atrophy-cerebellar variant (MSA-C). Cerebellar degeneration related to alcoholism and toxic causes were excluded because of lack of history and absence of cerebellar volume loss. Autoantibody screening and negative imaging results for primary or metastatic tumors further exclude a neoplastic etiology. Lack of abnormal signal changes in the posterior columns of the spinal cord and negative lab evaluation excludes vitamin B12 deficiency. No obvious clinical features, laboratory or imaging findings suggestive of chronic CNS infections such as HIV, CJD, Whipple disease, or neurosyphilis were present. Genetic screening for Friedreich ataxia and spinocerebellar ataxia was negative. FXTAS and MSA-C were excluded because of the absence of specific neuroimaging findings.
The neuroimaging findings of ECD can mimic multiple conditions, depending on the pattern of involvement, either meningeal or parenchymal. The meningeal pattern commonly presents with nodular meningeal thickening or mass lesion. Pathologies such as meningioma, lymphoma, tuberculosis, fungal infections, and sarcoidosis can have a similar appearance; however, these conditions can be excluded based on clinical, imaging, laboratory, and histopathologic analysis. In our patient, neuroimaging features were suggestive of parenchymal CNS involvement. The differential diagnoses may include parenchymal tumors such as glioma and lymphoma; however, lack of mass effect, edema, or rapid progression excludes these conditions. Middle cerebellar peduncle lesions can be seen in multiple sclerosis and other infectious-inflammatory demyelinating pathologies. However, the absence of corpus callosum or periventricular lesions and negative oligoclonal band excludes multiple sclerosis. The lack of leptomeningeal nodular enhancement and cranial neuropathies, and sparing of the hypothalamus excludes sarcoidosis.
Erdheim-Chester disease is an extremely uncommon, non-Langerhans form of histiocytosis of unknown etiology that can involve various organ systems including the musculoskeletal, cardiac, pulmonary, gastrointestinal, and central nervous systems. It equally affects both males and females in their fifth to seventh decades of life. Clinically, it can present as a focal asymptomatic process or multisystem fatal condition. Characteristic histiocytic organ infiltration often leads to non-specific manifestation of fever, malaise, and weight loss. The most frequent clinical systemic manifestation is skeletal involvement with bone pain. In ECD, the long bones of bilateral upper and lower extremities are commonly involved in a symmetric fashion. Plain film radiography of patients with ECD commonly demonstrates diffuse or patchy increased density, coarsened trabecular pattern, medullary sclerosis, and cortical thickening frequently affecting the meta-diaphysis of the long bones. Magnetic resonance imaging may reveal a geographic pattern of abnormal signal intensity in long bones, with relative sparing of epiphyses. Strong labeling of the distal ends of the long bones has been observed on 99mTc bone scintigraphs.
Extraskeletal manifestation is seen in approximately half of the patients with mass formation at sites such as the retroperitoneum, neck, orbit, or breast. Laboratory findings of ECD are non-specific and may include elevated SED, increased levels of alkaline phosphatase, and increased C-reactive protein levels. More specific laboratory findings may be demonstrated, depending on the physiologic functions affected by multisystemic ECD, and could help in understanding the patient’s distribution of the disease. Biopsy of long bones and other systemic sites involved in ECD typically demonstrates lymphocytes, macrophages, and lipid-laden histiocytes with immunohistochemistry positive for CD68 and negative for CD1a.
Central nervous system involvement is considered a poor prognostic factor and found in approximately one-third of patients with ECD, and can usually be associated with systemic disease. There are three patterns of CNS involvement in ECD: 1) a parenchymal infiltrative pattern with widespread lesions, nodules, or masses of the brainstem, cerebellum, and cerebral hemispheres (44% of patients); 2) a meningeal pattern with nodular thickening and meningioma-like lesions in approximately 37% of cases; and 3) a mixed pattern of parenchymal and meningeal involvement, which is seen in 19% of patients. Diabetes insipidus is the most common neurologic manifestation in ECD. Progressive cerebellar syndrome is the second most frequent CNS manifestation of ECD and can develop over several years. A variety of symptoms can be associated with brainstem and cerebellar involvement of ECD. These patients commonly present with slowly progressive cerebellar dysfunction displaying cerebellar dysmetria, hypermetric saccades, ataxia, dysarthria, nystagmus, dysdiadochokinesia, and pyramidal syndrome. Additionally, unlike most intracranial lesions, cerebellar lesions do not exhibit mass effect. The underlying pathogenesis is again similar to other systemic involvement with infiltration of intracranial tissue by foamy histiocytes with gliosis, extensive loss of myelin sheath, and marked sparing of axons. Magnetic resonance imaging is the modality of choice when evaluating the different CNS manifestations. On MRI, these lesions are characterized by high signal intensity on T2WI, low signal intensity on T1WI, and may show enhancement on contrast study. Cerebellar atrophy is rare and only visible in fewer percentages of patients with ECD.
Early diagnosis and recognition of ECD is important, as there is some evidence that treating ECD with interferon alpha may improve survival, as shown in a recent multicenter observational study. Erdheim-Chester disease should be regarded as a rare cause of adult-onset sporadic ataxia, especially when accompanied by enhancing pontine and cerebellar lesions with pathognomonic radiologic findings of sclerotic lesions and 99mTc uptake in the long bones. Extra-neurologic manifestations such as retroperitoneal fibrosis or orbital involvement are an additional clue for diagnosis. Biopsy of the amenable lesions is helpful for definitive diagnosis.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


