Fig. 1
Cyclized α-MSH analogs: a MTII, a seven-amino-acid derivative of α-MSH cyclized through lactamization of Asp2 and Lys7, and b ClickMTII, where the aspartic acid has been replaced by propargylglycine and the side-chain moiety of lysine has been changed to an azide, cyclized utilizing click chemistry between the terminal alkyne and azide to form a triazole
2 Experimental
2.1 Chemicals and Reagents
Fmoc-protected amino acids, 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, 1-hydroxybenzotriazole, benzotriazole-1-ly-oxy-tris-pyrrolidinophosphonium hexafluorophosphate, and Rink amide resin for peptide synthesis were obtained from Advanced ChemTech (Louisville, KY). DOTA-tris (t-Bu ester) was purchased from Macrocyclics (Dallas, TX). N,N-Dimethylformamide, N,N-diisopropylethylamine, methanol, trifluoroacetic acid, anisole, acetonitrile, PhSiH3, Pd(PPH3)4, and 2,6-lutidine were purchased from Thermo Fisher Scientific (Waltham, MA). Dichloromethane, triisopropylsilane, sodium ascorbate, copper(I) bromide, bovine serum albumin (BSA), 1,10-phenanthroline, HEPES, human serum, and citric acid were from Sigma Aldrich (St. Louis, MO). Dulbecco’s modified Eagle’s medium (DMEM), minimum essential medium (MEM), fetal bovine serum (FBS), and penicillin/streptomycin were from Invitrogen Life Technologies (Carlsbad, CA). B16/F10 murine melanoma cells were a gift from Dr. Michael Anderson at the University of Iowa (Iowa City, IA).
2.2 Peptide Synthesis
A total of ten linear and cyclized α-MSH peptide analogs were prepared, characterized, and tested in this study (Table 1). Four linear peptides, including α-MSH (Ac-SYSMEHFRWGKPV-NH2) and NDP-α-MSH (Ac-SYS-Nle-EHfRWGKPV-NH2), and their DOTA-conjugated variants, were synthesized on Rink amide resin at 0.1 mmol scale following standard Fmoc procedures on an AAPPTEC Apex 396 automated multiple peptide synthesizer. DOTA-tris (t-Bu ester) was coupled to the N-terminus of the protected linear peptides on-resin. Each peptide-resin (1 equiv peptide) was suspended in N,N-dimethylformamide (DMF), and DOTA-tris (t-Bu ester) (5 equiv), 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU; 5 equiv), 1-hydroxybenzotriazole (HOBt; 5 equiv), and N,N-diisopropylethylamine (DIPEA; 10 equiv) were added and the reaction mixed at 37°C overnight. Completeness of coupling was checked by the Kaiser test for free amines (Kaiser et al. 1970). The peptide-resin was filtered, washed with DMF, dichloromethane (DCM), and methanol, and allowed to dry. Peptides were cleaved and deprotected with 95:2.5:2.5 trifluoroacetic acid (TFA)/triisopropylsilane (TIS)/water for 2 h at room temperature, then precipitated with ice-cold ether. The peptides were purified on reversed-phase high-performance liquid chromatography (RP-HPLC) by injection onto a Vydac C18 semipreparative column (10 × 250 mm, 10 μm, 300 Å; Grace, Deerfield, IL) eluted at 5 ml/min with 0.1% TFA and a 20–30% gradient of acetonitrile over 30 min while monitoring absorbance at 280 nm. The peak of interest was collected, concentrated by rotary evaporation, and lyophilized. Purified peptides were characterized on an Agilent 1100 LC-ESI-ion trap mass spectrometer (Table 1).
Table 1
α-MSH peptide analogs with competitive binding assay and stability data
Peptide | Sequence | Mass (obs/calc) | IC50 (nM) | 24-h stability (% remaining) |
|---|---|---|---|---|
α-MSH | Ac-SYSMEHFRWGKPV-NH2 | 1,664.7/1,664.9 | 2.1 ± 0.4 | N.D. |
DOTA-α-MSH | DOTA-SYSMEHFRWGKPV-NH2 | 2,009.0/2,009.3 | 1.8 ± 0.3 | 7.4 |
NDP-α-MSH | Ac-SYS-Nle-EHfRWGKPV-NH2 | 1,646.6/1,646.9 | 0.21 ± 0.03 | N.D. |
DOTA-NDP-α-MSH | DOTA-SYS-Nle-EHfRWGKPV-NH2 | 1,990.6/1,991.2 | 0.59 ± 0.11 | 41 |
MTII | Ac-Nle-cyclo[DHfRWK]-NH2 | 1,024.6/1,024.2 | 0.26 ± 0.08 | N.D. |
ClickMTII | Ac-Nle-cyclo[Pra-HfRW-Lys(N3)]-NH2 | 1,048.3/1,048.2 | 0.39 ± 0.06 | N.D. |
DOTA-ClickMTII | DOTA-Nle-cyclo[Pra-HfRW-Lys(N3)]-NH2 | 1,392.3/1,392.6 | 25 ± 1 | 100 |
DOTA-GG-ClickMTII | DOTA-GG-Nle-cyclo[Pra-HfRW-Lys(N3)]-NH2 | 1,506.1/1,506.7 | 2.7 ± 0.8 | 99 |
ClickMTII-K-DOTA | Ac-Nle-cyclo[Pra-HfRW-Lys(N3)]-K(DOTA)-NH2 | 1,562.4/1,562.8 | 1.4 ± 0.2 | 99 |
ClickMTII-GGK-DOTA | Ac-Nle-cyclo[Pra-HfRW-Lys(N3)]-GGK(DOTA)-NH2 | 1,676.6/1,676.9 | 1.1 ± 0.2 | 100 |
A single lactam-bridge-cyclized peptide (MTII, Ac-Nle-cyclo[DHfRWK]-NH2) was synthesized as described above, with the exception that Fmoc-Asp(Allyl)-OH and Fmoc-Lys(Alloc)-OH residues were inserted for selective deprotection to facilitate cyclization of the Fmoc-protected peptide on-resin (Fig. 1). Following synthesis, according to published procedures, the Allyl and Alloc groups were to be removed selectively by adding PhSiH3 (24 equiv) and Pd(PPH3)4 (0.10 equiv) in DCM and mixing for 10 min at room temperature (Thieriet et al. 1997). The peptide-resin was filtered and washed with DCM, and the deprotection was repeated. Complete removal of the Alloc group was tested by running the Kaiser test to detect free amine of the lysine side-chain. Peptide cyclization between the acid group of the aspartic acid and the primary amine of the lysine was achieved by addition of benzotriazole-1-ly-oxy-tris-pyrrolidinophosphonium hexafluorophosphate (PyBOP; 1.1 equiv), DIPEA (2.2 equiv), and HOBt (0.5 equiv) in DMF. The reaction was mixed overnight at room temperature, and the resin was filtered and washed with DMF, DCM, and methanol. The cyclized peptide was cleaved and deprotected with 92:4:2:2 TFA/TIS/water/anisole for 2 h at room temperature, then precipitated with ice-cold ether. The cyclic MTII peptide was purified by semipreparative HPLC as described above using a 20–35% gradient of acetonitrile, and the purified peptide was characterized on an Agilent 1100 LC-ESI-ion trap mass spectrometer.
Five click-cyclized MTII analogs were synthesized by similar methods (Table 1; Fig. 1). In this case, the internal amino acid sequences for these five MTII analogs differed, with the insertion of of Pra and Lys(N3) residues to facilitate cyclization through triazole ring formation via “click” chemistry. In addition, an Fmoc-Lys(Dde)-OH was added for peptides with a C-terminal lysine residue. Following synthesis, peptides containing a Lys(Dde) were selectively deprotected by treatment with 2% hydrazine for 15 min. Peptides were cyclized by modification of methods by Turner et al. (2007). Briefly, the peptide-resin was swelled in DMF and DIPEA prior to addition of 2,6-lutidine (10 equiv each). Sodium ascorbate was added as 1% solution in 1:4 water/DMF (v/v; 3 equiv) followed by copper(I) bromide as 1% solution in acetonitrile (1 equiv). Sodium ascorbate was added to maintain copper as the monovalent, catalytically active cationic form. The reaction mixture was stirred at 37°C overnight, and the peptide-resin was filtered and washed with DMF, DCM, and methanol. DOTA-tris (t-Bu ester) was coupled to the primary amine (either N-terminus or lysine side-chain), and peptides were cleaved/deprotected as described above. To examine the effect of DOTA conjugation on binding affinity and stability, four ClickMTII variants were constructed with DOTA conjugated to either the N– or C-terminus of the peptide: (1) DOTA coupled directly to the free amine of the N-terminus, (2) DOTA coupled through a glycine–glycine (GG) spacer added to the N-terminus of the peptide sequence, (3) DOTA attached to the C-terminus via the ε-amine side-chain of a lysine residue added to the C-terminal end of the peptide, or (4) through the lysine side-chain of a glycine–glycine–lysine spacer. The click-cyclized peptides were purified by semipreparative HPLC using a 20–30% or 20–35% gradient of acetonitrile, and the purified peptides were characterized on an Agilent 1100 LC-ESI-ion trap mass spectrometer (Table 1).
2.3 In Vitro Competitive Binding Assay
B16/F10 murine melanoma cells were cultured in high-glucose DMEM supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in 150-cm3 culture flasks at 37°C in a humidified 5% CO2 incubator; cells were grown to 70% confluency and used in competitive binding assays similar to the methods developed by Siegrist et al. (1988). Cells were scraped from the culture flasks, counted on a Beckman Coulter cell counter, and resuspended in binding media (MEM containing 25 mM HEPES, 0.2% BSA, and 0.3 mM 1,10-phenanthroline). For binding assays, a total volume of 500 μL was used in 1.5-mL Eppendorf tubes. The cell suspension (50 μL containing approximately 4 million cells) was incubated with approximately 30,000 cpm of [125I]-[Nle4,d-Phe7]-α-MSH and increasing concentrations of α-MSH or peptide analog ranging from 10−6 to 10−11 M. The tubes were mixed at 25°C, 600 rpm on an Eppendorf Thermomixer for 1.5 h. Following incubation, cells were pelleted by centrifugation, the media was aspirated, pellets were transferred to 12 × 75 mm glass tubes, and the radioactivity of the cell pellet was determined using a PerkinElmer Cobra II gamma counter (PerkinElmer, Freemont, CA). All handling of radioactive materials was conducted using procedures and protocols approved through the University of Iowa Environmental Health and Safety Committee and Health Protection Office, adhering to as low as reasonably achievable (ALARA) principles. Each experiment was performed in quadruplicate, and binding curves were plotted; 50% inhibition concentration (IC50) values and their associated standard errors were calculated with GraphPad Prism 5 curve-fitting software (GraphPad Prism version 5.01 for Windows; GraphPad Software, San Diego, CA).
2.4 Radiochemistry
64Cu was obtained as 64CuCl2 solution from the College of Medicine, Washington University in Saint Louis (WashU, St. Louis, MO, USA). Radiolabeling of DOTA-peptides with 64Cu was carried out as described previously (Anderson et al. 2008; Boswell et al. 2008; Brechbiel 2008; Hausner et al. 2009). Briefly, 64Cu was received from WashU in approximately 10–40 μl of mildly acidic solution in a screw-top plastic “v-vial,” enclosed within appropriate lead shielding. To this vial was added ultrapure water or pH 6 sodium acetate buffer solution to final volume of 200 μl, and the solution was mixed. The 200 μl solution was withdrawn and transferred to a 1.5-ml plastic v-vial (SealRite, natural microcentrifuge tube, Cat# 1615-5500, USA Scientific), and a measurement of the total activity was determined by dose calibrator (CRC-25R, Capintech, Ramsey, NJ). Aliquots of appropriate radioactivity amounts could then be transferred volumetrically to appropriate radiolabeling solutions. Generally, 10 nmoles peptide was suspended in 50 μl pH 6 acetate buffer. To this solution was added an appropriate aliquot of 64Cu master solution, and the solution was incubated for 45 min at 70°C. This solution was used without further purification for in vitro stability testing. The identity of radiolabeled species was confirmed by radioHPLC retention time comparison with UV traces of the unlabeled species at 280 nm.
Radiolabeling with 68Ga was carried out through the use of a 68Ge/68Ga generator system (IGG100; Eckert Ziegler GmbH, Berlin, Germany) with total 68Ge activity of approximately 600 MBq at the time of experiments presented here. The system was aged between 5–10 months for experiments conducted for this investigation. The methodology employed involves a single-step purification of 68Ga eluted from the generator with 10 ml 0.1 M HCl at flow rate of approximately 2 ml/min directly to a cation exchange (Strata™-XC, 33 μm, Strong Cation, 30 mg/ml #8B-S029-TAK, Phenomenex) column mounted by supports fabricated in house. Following generator elution, the generator outflow line is disconnected and the Strata-XC column is air-dried using a 20-ml plastic syringe (2x). Once dry, pure 68Ga is eluted from the Strata-XC column with 400 μl of 98% acetone/0.05 M HCl (prepared weekly and stored at 4°C) directly to a glass vial containing 5–10 nmoles DOTA-peptide conjugate dissolved in 5 ml pure water that had been preheated to approximately 80°C. This solution was then heated (open vessel) to 100°C and 68Ga (with the DOTA-peptide) are incubated for 15 m. Acetone is evaporated to less than 300 ppm in the final purified drug product by this method. Following the radiolabeling incubation period, the reaction mixture containing the [68Ga]-peptide and a small amount of free 68Ga was drawn up through a Strata™-X cartridge (33 μm Polymeric Reversed Phase C-18, 30 mg/1ml, #8B-S100-TAK, Phenomenex® Inc., Torrance, CA, USA) that had been preconditioned by passing 1 ml 95% ethanol (USP for injection) and 2.5 ml pure water. The cartridge was then rinsed with 2 ml water to remove any residual free 68Ga, and finally the purified 68Ga-peptide conjugate was eluted in 500 μl of 1:1 ethanol (95%):isotonic saline solution. Radiochemical purity was determined by radioHPLC as described for 64Cu.
2.5 Serum Stability Assay
Stability of chelator-modified peptide bioconjugates was determined by radioTLC (64Cu labeled) using iTLC-SG glass microfiber chromatography paper impregnated with silica gel (Varian, Lake Forest, CA, USA) as described in detail previously.(Rockey et al. 2011) Briefly, iTLC paper was cut into 5 × 25 cm2 strips and heated at 100°C for at least 30 min prior to measurements. Radiolabeled peptides were added to human serum and allowed to incubate at 37°C. At specified time points, 2 μl of the labeled peptide/serum mixture was spotted at a predetermined origin on the strips, and the iTLC strips were developed for about 3 min with a mobile phase of 0.1 M citric acid. Under these conditions, it was determined in advance that stable (intact) peptide remains at the origin, while free 64Cu migrates with the mobile phase and peptide fragmentation can be observed by intermediately dispersed retention times. The strips were then dried with a common hairdryer. Possible fragmentation of the radiolabeled chelator-modified bioconjugates was determined through exposure of the iTLC strips to a storage phosphor screen (Fujifilm, Minamiashigara, Kanagawa, Japan) and subsequent screen imaging with a Typhoon™ FLA 7000 PhosphorImager (GE Healthcare Bio-sciences AB, Uppsala, Sweden). Data analysis of the phosphor images was performed using ImageQuant™ Analysis Toolbox software (TL 7.0; GE Healthcare Bio-sciences AB, Uppsala, Sweden). The percent intact radiolabeled chelator-modified peptides was calculated by dividing the integrated signal intensity over the bottom half of the TLC strips by the total integrated signal intensity over the entire length of the TLC strips. The technique was confirmed by radioHPLC. The pH of solutions was confirmed by use of an Horiba Compact pH-meter (model B-213; Horiba Scientific Instruments, Ann Arbor, MI, USA), calibrated daily at pH 4 and pH 7 using standard buffers or colorpHast pH paper (0–14 range; Merck KGaA, Darmstadt, Germany).
2.6 In Vivo PET Imaging and Biodistribution Studies
All animal experiments were performed according to approved protocols that were compliant to all rules and regulations of federal regulatory bodies and the University of Iowa Animal Care and Use Committee.
Female SCID hairless mice (6–8 weeks old) were implanted subcutaneously with three million MC1R-expressing cells (B16-F10 murine melanoma). Five to seven days after inoculation, tumors had grown to approximately 100–200 mg and the tumor-bearing mice were subjected to in vivo biodistribution and imaging studies using 68Ga-labeled MTII variants. For biodistribution studies, approximately 7.5 MBq (200 μCi) of [68Ga]DOTA-MTII variants (specific activity 30–50 MBq nmole−1 and radiochemical purity >98%) was injected intravenously into the lateral tail vein using tuberculin insulin-type syringes (Becton Dickinson; 8 mm (5/16”), 500 μl, 31G Ultra-Fine™ needle) (Guo et al. 2010). Following injection of the radiolabeled tracer, animals were placed back in cages that were lined with absorbent material to sequester excretion for measurement at the conclusion of the uptake period (1.5 h). The uptake period for the comparison was determined by consideration of the radionuclide half-life of 68Ga, as well as preliminary pharmacokinetic studies using [68Ga]DOTA-Amide-NDP-α-MSH and literature reviews of other cyclized α-MSH analogs. At the conclusion of the uptake period, animals were sacrificed by approved procedures. Blood, tissues, and organs of interest (heart, liver, kidneys, lung, brain, muscle, and tumors) were resected, dabbed on blotter paper to remove excess blood, transferred to preweighed snap-cap v-vials, weighed, and assayed for radioactivity by scintillation counting. The radioactivity in excretion (urine and feces) was determined by the total activity that could be wiped from the floor of wire-bottom cages using the inserted absorbent pads and urine extracted from the bladder. Following resections and removal of urine from the bladder, the carcass was also counted by dose calibrator measurement to complete a measure of radioactivity balance. The radioactivity measurements were normalized to percent injected dose per gram (% ID/g) of tissue for each tissue sample with appropriate decay corrections applied. An evaluation of the biodistribution of “free” 68Ga3+ was conducted to determine the possible contribution of free radio metal to tumor accumulation of injected dose. Finally, receptor-mediated tumor accumulation to xenograft tumors was confirmed by co-injection of 50–750 μg unlabeled NDP-α-MSH together with 68Ga DOTA-peptide conjugates.
In vivo imaging studies were conducted to complete the preliminary evaluation of the click-cyclized peptides. For imaging, animals were anesthetized and injected with approximately 25 MBq [68Ga]-peptide (specific activity 30–50 MBq nmole−1) suspended in approximately 100 μl saline solution. Animals were allowed access to food and water at all times. Imaging of the biodistribution of the radiolabeled peptide was conducted at 1.5 h post injection (p.i.) using an Inveon™ small-animal PET/CT (Siemens, Inc., Knoxville, TN, USA). A 15-min static scan was employed for imaging. Immediately prior to scanning, the bladder contents were carefully expunged and collected in an absorbent pad for quantification of urine radioactivity and to prevent urinary contamination of the imaging bed.
3 Results
3.1 Peptide Synthesis
The lactam-bridge-cyclized α-MSH peptide analog MTII and click-cyclized variant ClickMTII were synthesized utilizing similar Fmoc procedures (Rink amide resin) and final Fmoc removal and N-terminal acylation. Thus, the molecular compositions were identical, with the exception of the linkage used for cyclization (Fig. 1). For lactam bridge formation of MTII, removal of allyl/alloc protecting groups was required to prepare the peptide for cyclization by amide bond formation via the acid moiety of the aspartic acid and the primary amine of lysine (lactam bridge formation). While much of the synthetic strategy for preparation of the ClickMTII derivative was the same, the cyclization strategy in this case was achieved through cycloaddition of the alkyne group of propargylglycine and the azide of an azido-modified lysine residue to form the triazole ring (Fig. 1). Following cyclization, peptides were deprotected and cleaved from the resin, purified by semipreparative HPLC, and characterized by LC–MS (data not shown).
To facilitate radiolabeling with 64Cu and 68Ga, a DOTA chelator was conjugated to the MTII and ClickMTII variants by standard methods. To conjugate DOTA to the N-terminus of MTII, the N-terminal Fmoc was left in place at the end of the peptide synthesis. The peptide was then treated with palladium catalyst and phenyltrihydrosilane for removal of the allyl and alloc protecting groups and cyclized as previously described (see Sect. 2.2, “Peptide Synthesis”). However, HPLC analysis of the N-terminal Fmoc-protected MTII peptide before cyclization by HPLC showed the presence of two major peaks, corresponding to the peptide with and without the N-terminal Fmoc (data not shown). Thus, difficulty in synthesizing an N-terminal Fmoc-protected MTII prevented smooth attachment of DOTA to this peptide, and alternative click chemistry strategies were pursued.
On the other hand, ClickMTII was synthesized smoothly without N-terminal acylation, facilitating DOTA addition to the free amine of the N-terminus. In this case, N-terminal Fmoc protection was not necessary as cyclization was carried out by click chemistry, which does not involve a primary amine. DOTA was then coupled to the peptide by formation of an amide bond between a carboxylic acid on DOTA and the primary amine of the N-terminus. DOTA-ClickMTII was finally deprotected and cleaved from the resin, purified by semipreparative HPLC, and characterized by LC–MS (Table 1).
In addition to the MTII and ClickMTII peptides, native α-MSH, NDP-α-MSH, and their N-terminal DOTA derivatives were prepared for use in in vitro experiments for structure–activity comparisons (Table 1). Preparative HPLC-purified peptides were rechromatographed on analytical RP-HPLC to establish structure on the basis of retention time and mass determined by LC–MS. For each purified peptide, a single major peak demonstrated >95% purity that also produced a singly charged positive ion in the ESI–MS corresponding to the calculated mass of the desired peptide (Table 1).
3.2 Radiolabeling
DOTA-GG-ClickMTII and ClickMTII-GGK-DOTA were radiolabeled with 64Cu in acetate buffer (pH 6) for 45 min at 70°C. For 64Cu, no further purification was required to obtain radiochemical purity of >98% with specific activity of >35 MBq nmole−1 peptide conjugate (FIG). Similar results were obtained for radiolabeling with 68Ga. In this case, radiolabeling was conducted by elution of 68Ga from a manual generator system. Following a radiolabeling step (5 ml water, pH 3.5, 15 min, 100°C), final purification of the 68Ga-labeled peptides was performed using a disposable C18 reverse-phase cartridge. Radiochemical purity was routinely >98% with specific activities up to 45 MBq nmole−1 of peptide conjugate.
3.3 In Vitro Competitive Binding Assays
The ClickMTII peptide analogs, without and with DOTA, were evaluated for their binding affinity to melanocortin receptors in B16/F10 murine melanoma cells. Competitive binding assays were carried out using the radiolabeled ligand [125I]-NDP-α-MSH and increasing amounts of α-MSH analog. Experiments were carried out in quadruplicate, and binding curves were plotted as the mean and standard error of the four sets of data (Fig. 2; Table 1). The uncertainties of these results are based on the standard deviation of the regression curve fit-derived affinity results of the quadruplicate measurements. MTII (closed circles) and ClickMTII (open circles) give similar curves, with IC50 values of 0.26 and 0.39 nM, respectively. Addition of DOTA to the N-terminus of ClickMTII (DOTA-ClickMTII; closed triangles) results in a 64-fold decrease in binding affinity and an IC50 of 25 nM. Due to the large increase in the IC50 with the addition of DOTA, three other analogs were synthesized to optimize the placement of DOTA. DOTA was conjugated to the C-terminus through the side-chain of an added lysine residue (ClickMTII-K-DOTA; IC50 = 1.4 nM). Two peptides were also synthesized with glycine spacers to see if the added distance between the DOTA and the peptide would increase binding affinity (DOTA-GG-ClickMTII and ClickMTII-GGK-DOTA; IC50 = 2.7 and 1.1 nM, respectively). Binding affinity data are summarized in Table 1.
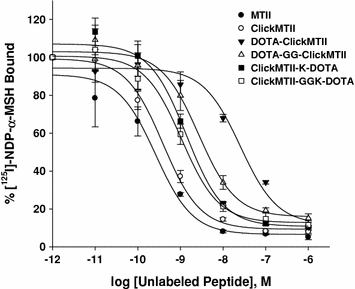
Fig. 2
Competitive inhibition of 125I-NDP-α-MSH binding to B16/F10 murine melanocytes by α-MSH analogs. Competitive binding curves are the mean and standard error of quadruplicate experiments
3.4 Serum Stability
DOTA-conjugated peptides were radiolabeled with 64Cu in pH 6.0 acetate buffer and used without further purification. Labeled peptides were added to human serum and incubated at 37°C to determine both the stability of the radionuclide within the DOTA complex and the stability of the peptide to serum proteases. At specified time points, samples were removed and analyzed by radioTLC as described in the experimental procedures. When spotted on the iTLC strip, intact [64Cu]-DOTA-peptide remained where initially spotted, while free 64Cu and peptide fragments migrated with variable retention characteristics with the solvent. Immediately upon addition of the peptides to the serum, a reference sample was taken of each peptide. At this zero time point, all [64Cu]-DOTA-peptides remained intact and did not migrate with the solvent (Fig. 3). Analysis of the iTLC strips at 24 h showed that a significant amount of both linear test peptides (DOTA-α-MSH and DOTA-NDP-α-MSH) were degraded by the serum, while the cyclized DOTA-ClickMTII analogs were intact (Fig. 3). The four DOTA-ClickMTII peptides remained stable throughout the entire 24 h experiment, while the linear DOTA-α-MSH and DOTA-NDP-α-MSH begin to degrade after 1 h in serum. By 24 h, only 7.4% of DOTA-α-MSH and 41% of NDP-α-MSH remained, while >99% of all four DOTA-conjugated cyclized ClickMTII analogs remained stable (Fig. 3; Table 1).
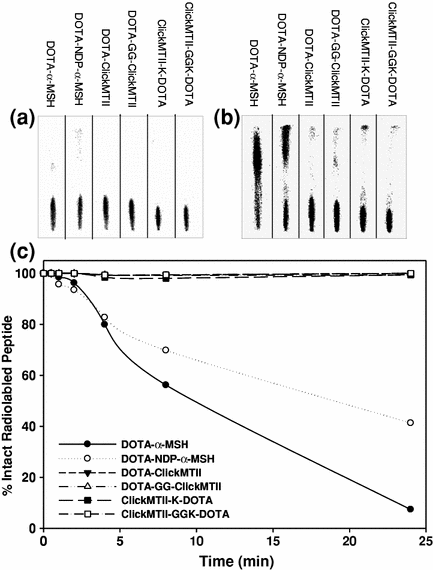
Fig. 3
 via Cation Exchange Cartridges: Versatile Options
via Cation Exchange Cartridges: Versatile Options
 Experience in Peptide Receptor Radionuclide Therapy
Experience in Peptide Receptor Radionuclide Therapy
 Intermediate-Affinity Monoclonal Antibodies Targeting EGFR and HER2/c-neu: Preparation and Preclinical Evaluation
Intermediate-Affinity Monoclonal Antibodies Targeting EGFR and HER2/c-neu: Preparation and Preclinical Evaluation
 Novel 68Ga-Labeled Pteroic Acid-Based PET Tracer for Tumor Imaging via the Folate Receptor
Novel 68Ga-Labeled Pteroic Acid-Based PET Tracer for Tumor Imaging via the Folate Receptor
 Rare Case of a Large Spinal Meningioma with Mediastinal Extension and Malignant Behavior Classified Histologically as Benign
Rare Case of a Large Spinal Meningioma with Mediastinal Extension and Malignant Behavior Classified Histologically as Benign
 Role of 68Ga-Labeled Somatostatin Analogs in the Workup of Patients with NETs: AIIMS Experience
Role of 68Ga-Labeled Somatostatin Analogs in the Workup of Patients with NETs: AIIMS Experience




Stability of [64Cu]-DOTA-peptide analogs in human serum. Radiolabeled peptides were incubated in human serum at 37°C, and at specified time points samples were spotted on iTLC paper and developed with 0.1 M citric acid. a shows the peptides in serum at t = 0 min; all of the radiolabeled peptide remains at the bottom of the plate. By 24 h, the linear peptides α-MSH and NDP-α-MSH are degraded by the serum while the cyclic ClickMTII analogs remain intact (b). c graphically represents the degradation data over the 24 h time period for all peptides
Related posts:
via Cation Exchange Cartridges: Versatile Options
Experience in Peptide Receptor Radionuclide Therapy
Intermediate-Affinity Monoclonal Antibodies Targeting EGFR and HER2/c-neu: Preparation and Preclinical Evaluation
Novel 68Ga-Labeled Pteroic Acid-Based PET Tracer for Tumor Imaging via the Folate Receptor
Rare Case of a Large Spinal Meningioma with Mediastinal Extension and Malignant Behavior Classified Histologically as Benign
Role of 68Ga-Labeled Somatostatin Analogs in the Workup of Patients with NETs: AIIMS Experience
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
