Fig. 8.1
Sodium channel blocking agents improve neurological status of rodents with EAE. (a, b) Clinical scores (mean ± SEM) are shown for untreated C57/Bl6 mice with progressive EAE (inverted triangles in a and filled diamonds in b); both phenytoin (filled boxes in a) and carbamazepine (filled circles in b) treatment of mice with progressive EAE significantly improved the clinical course of the disease. Oral administration of phenytoin or carbamazepine was started on day 10 following MOG inoculation, as indicated by the horizontal bar, and continued until the end of the study at day 28. (c) The progression of chronic-relapsing EAE in DA rats followed a relapsing disease course (filled circles). Flecainide treatment commencing at 7 days post spinal cord homogenate inoculation reduced the severity of the disease course (filled circles) (modified and reprinted with permission from Lo et al. 2003, Bechtold et al. 2004, Black et al. 2007)
In both progressive (C57/Bl6 mice) and chronic-relapsing (DA rats) forms of rodent EAE, administration of sodium channel blocking agents significantly improved the clinical course of the disease (Lo et al. 2003; Black et al. 2006, 2007; Bechtold et al. 2002, 2004, 2006). Treatment of C57/Bl6 mice with phenytoin or carbamazepine commencing on day 10 following MOG injection (using an oral treatment that resulted in phenytoin and carbamazepine serum levels within the human therapeutic range) significantly improved the clinical course in the treated mice compared with untreated mice, as assessed from day 13 until the end of the study at day 28 (Fig. 8.1a, b; Lo et al. 2003; Black et al. 2007). Likewise, flecainide administered to DA rats, with EAE by twice daily injections beginning either 7 days following (Fig. 8.1c) or 3 days prior to inoculation, significantly improved the mean peak deficit and terminal deficit scores compared with vehicle-treated rats (Bechtold et al. 2004). Lamotrigine treatment in DA rats provided more limited clinical improvement in the acute course of EAE compared with untreated rats but significantly improved the terminal deficit in the lamotrigine-treated rats in comparison with vehicle-treated EAE rats (Bechtold et al. 2006).
These studies demonstrated the efficacy of sodium channel blockade as an intervention that improves the neurological status in EAE for the duration of drug administration (up to 28 days) following induction of EAE. Subsequently, it was shown that phenytoin administration significantly improved the clinical course of EAE in C57/Bl6 mice when administered for up to 180 days, which was the endpoint of this study (Black et al. 2006). Taken together, these observations clearly established sodium channel blocking agents as providing significant improvement in the neurological status of rodent EAE and suggested their use as therapeutic agents in neuroinflammatory disorders.
8.2.2 Axonal Protection
Early studies in EAE demonstrated a loss of axons (Raine and Cross 1989; White et al. 1992; Kornek et al. 2000; Onuki et al. 2001; Wujek et al. 2002), which was correlated with the accumulation of nonremitting disability (Wujek et al. 2002). To determine whether the improved neurological status observed in EAE with sodium channel blocking agents was accompanied by protection of axons from degeneration, we assessed the magnitude of axonal degeneration in spinal cords of treated versus untreated mice and rats with EAE. Our results with the two distinct EAE models, and with three different sodium channel blocking agents, demonstrated consistent protection of axons in these animals. In C57/Bl6 mice, untreated EAE was accompanied by significant loss of axons within the dorsal corticospinal tract (CST, a descending tract) and the dorsal funiculus (DF, ascending tracts) (Fig. 8.2a; Black et al. 2007), with an approximately 60% loss of axons in the CST and a 40% loss in the DF compared to control mice, as assessed at 28–30 days post-MOG inoculation (Fig. 8.2a; Lo et al. 2003). Interestingly, in a separate study, axonal loss in the CST and DF at 180 days post-MOG inoculation were similar to those at 28–30 days post-MOG injection (Black et al. 2007), indicating that axonal degeneration in this murine EAE model occurs primarily during the initial phase of the disease. In contrast to untreated mice with EAE, treatment with phenytoin at clinically relevant levels significantly attenuated the loss of axons in mice with EAE at 28–30 days post-MOG inoculation, such that there was only a 28% loss of axons in the corticospinal tract and a 17% loss in the dorsal funiculus compared with control values (Fig. 8.2a). Significantly, the axonal protection afforded by phenytoin treatment persisted during longer treatment periods, for at least 180 days (Black et al. 2007).
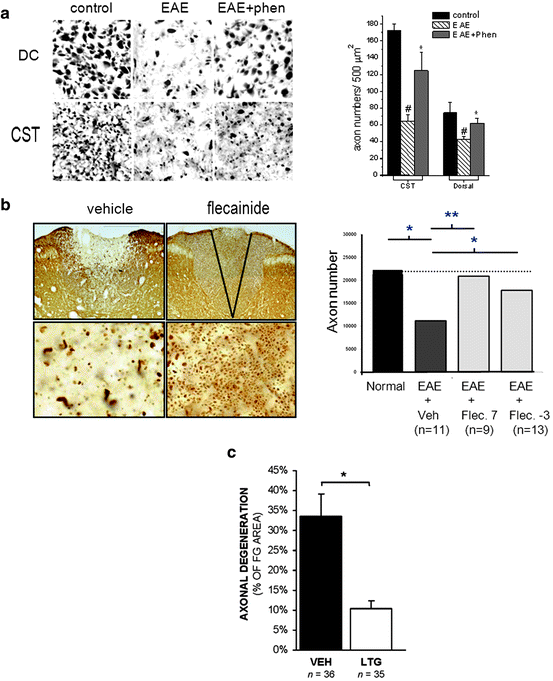
Fig. 8.2
Sodium channel blocking agents reduce the loss of spinal cord axons in EAE. (a) There was a substantial loss of axons in the dorsal funiculus (DF) and dorsal corticospinal tract (CST) in untreated EAE compared to control mice. Phenytoin-treated mice with EAE (EAE + phen) exhibited substantial increases in axons in the DF and CST compared to untreated EAE mice. Quantification of CST and DC axons (axon numbers/500 μm2) from control, untreated EAE, and phenytoin-treated EAE mice demonstrates a significant (p < 0.05) reduction of axon counts in the CST and DC in untreated EAE (hash symbols) compared to untreated controls. Phenytoin treatment of EAE (asterisks) results in a significant (p < 0.05) increase in axons within the CST and DC compared to untreated EAE. (b) Sections of dorsal columns immunolabeled with neurofilament showed substantial loss of axons in vehicle-treated DA rats with EAE. In comparison, flecainide administration commencing at day 7 postinoculation greatly reduced the loss of axons. Quantification of the number of axon within a selected portion of the dorsal columns Fig. 8.2 (continued) (shown by the “V” in the upper right illustration) of normal, vehicle-treated EAE, and EAE rats treated with flecainide commencing at day 7 or -3 demonstrates a significant loss of axons in untreated EAE dorsal columns, with significant protection of axons mediated by flecainide therapy. *p < 0.05, **p < 0.01. (c) Histogram showing the magnitude of axonal degeneration in severely diseased DA rats with chronic-relapsing EAE. Lamotrigine treatment significantly reduced axonal degeneration in DA rats with CR-EAE compared with vehicle-treated rats with EAE (*p < 0.050) (modified and reprinted with permission from Lo et al. 2003, Bechtold et al. 2004, 2006, Black et al. 2007)
Similar protective effects of sodium channel blockade were seen in DA rats with chronic-relapsing EAE treated with flecainide or lamotrigine. DA rats inoculated with spinal cord homogenate exhibited prominent axonal loss and ongoing axonal degeneration, particularly within the medial region of the dorsal columns (fasciculus gracilis) (Fig. 8.2b, c; Bechtold et al. 2004), that was closely associated with the peak deficit scores exhibited by these rats. DA rats with EAE that had peak scores ≥8 (on 15-point scale) displayed an approximately 40% loss of axons relative to control rats. In contrast, both flecainide (Bechtold et al. 2002, 2004) and lamotrigine (Bechtold et al. 2006) treatments provided significant protection of spinal cord axons when administered to DA rats with EAE (Fig. 8.2b, c). In comparison with the number of axons in normal rats, flecainide treatment commencing either 3 days prior to or 7 days following homogenate injection provided near complete axonal protection, with axonal counts remaining at 98% ± 20% and 83% ± 14% of control values, respectively (Bechtold et al. 2004). Similar results were obtained with lamotrigine treatment of DA rats with EAE, such that, when assaying degeneration as a percentage of total area measured, lamotrigine-treated rats exhibited axonal degeneration in only 9% of the area, while untreated rats displayed degeneration in 31% of the area (Bechtold et al. 2006). The circulating concentration of lamotrigine (mean 11.5 μg/ml) in EAE was within the clinical therapeutic range (1–15 μg/ml) for the treatment of epilepsy. Flecainide therapy was also found to be effective in axonal protection in rats with a related disease, namely, experimental autoimmune neuritis, a model of Guillain–Barré syndrome (Bechtold et al. 2005).
8.2.3 Functional Protection of Axons
To assess whether the preservation of axons observed with morphological methods in EAE with sodium channel blocking therapy was accompanied by preservation of function in the axons, we performed recordings of the compound action potential (CAP) from the dorsal surface of spinal cords of untreated and sodium channel blocker-treated mice and rats with EAE. Our results in both rodent species with EAE demonstrated that treatment with sodium channel blocking agents provides increased numbers of functional axons (greater CAPs) compared with untreated EAE, consistent with the attenuation of axonal loss and improvement of clinical status in these animals.
C57/Bl6 mice with EAE displayed clear improvements in CAP areas when administered phenytoin compared with untreated mice with EAE. The maximal CAP area in untreated mice with EAE was significantly attenuated compared with control mice (0.038 mV ms in EAE vs. 0.16 mV ms in controls; Fig. 8.3a; Lo et al. 2003). In contrast, phenytoin-treated mice with EAE exhibited maximal CAP areas (0.13 mV ms) that were significantly greater than those in untreated mice with EAE (0.038 mV ms) and that approached control values (0.16 mV ms). In addition, assessment of mean conduction velocities demonstrated significant improvement in conduction in phenytoin-treated versus untreated mice with EAE (control: 11.6 ± 2.1 m/s; EAE: 2.3 ± 1.7 m/s; EAE + phenytoin: 13.2 ± 1.1 m/s; Lo et al. 2003).
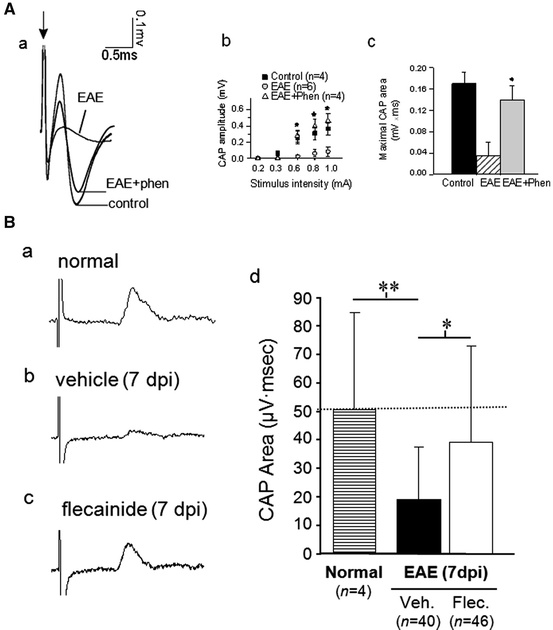
Fig. 8.3
Electrophysiological assessment of dorsal spinal cord axonal function in rodent EAE. (A) (a) Superimposed CAP traces from representative control, untreated EAE, and phenytoin-treated C57/Bl6 mice with EAE. The typical biphasic wave seen in controls is highly attenuated in untreated EAE but is restored in phenytoin-treated EAE. (b) Mean CAP amplitudes (±SEM, mV) in control, untreated EAE, and phenytoin-treated EAE at different stimulating current intensities (*p < 0.01, phenytoin-treated EAE compared to untreated EAE). (c) Mean supramaximal CAP area (±SEM, mV × ms) in control, untreated EAE, and phenytoin-treated EAE (*p < 0.05 phenytoin-treated EAE compared to untreated EAE). (B) Representative records from normal (a) and CR-EAE DA rats treated with vehicle (b) or flecainide (c) from 7 days postinoculation. (d) The area of the CAP was significantly reduced from normal, but this reduction was largely prevented by flecainide treatment. This protection was statistically significant with therapy from 7 dpi (*p < 0.05, **p < 0.01) (modified and reprinted with permission from Lo et al. 2003, Bechtold et al. 2004)
A similar protective effect on axonal conduction was seen in DA rats with chronic-relapsing EAE. Normal DA rats exhibited supramaximal-stimulated monophasic CAP areas of around 50–55 μV ms (Fig. 8.3b; Bechtold et al. 2004, 2006). The CAP areas of DA rats with EAE were significantly reduced by 40–60% (20–30 μV ms). Consistent with the observed preservation of axons achieved with treatment with sodium channel blocking agents, both flecainide (Bechtold et al. 2002, 2004) and lamotrigine (Bechtold et al. 2006) administration to DA rats with EAE significantly increased the CAP areas compared with vehicle-treated DA rats with EAE (Fig. 8.3b). Flecainide treatment commencing either-3 days prior to or 7 days following spinal cord homogenate inoculation significantly improved the CAP areas (-3-day flecainide: 28 μV ms vs. untreated 21 μV ms; 7-day flecainide: 37 μV ms vs. untreated 20 μV ms; Bechtold et al. 2004). Lamotrigine also preserved axonal conduction in DA rats with EAE, increasing the CAP area to 43 μV ms compared with 31 μV ms for untreated rats (Bechtold et al. 2006).
8.2.4 Inflammatory Infiltrates
The onset and progression of EAE is associated with infiltration of immune cells into the CNS (Zamvil and Steinman 1990; Gold et al. 2006). Interestingly, sodium channels are expressed by immune cells, including T lymphocytes (Gallin 1991; Lai et al. 2000; Fraser et al. 2004), microglia (Korotzer and Cotman 1992; Norenberg et al. 1994; Eder 1998; Craner et al. 2005; Black et al. 2009) and macrophages (Carrithers et al. 2007). Sodium currents are suggested to contribute to T lymphocyte activation and co-stimulation (Khan and Poisson 1999; Lai et al. 2000), while phagocytosis by microglia and macrophages and migration by microglia in vitro are attenuated by sodium channel blockade (Craner et al. 2005; Carrithers et al. 2007; Black et al. 2009). Figure 8.4 shows a significant reduction in phagocytosis by LPS-activated microglia in vitro when treated with either TTX or phenytoin (Black et al. 2009). Carrithers et al. (2007, 2009) showed that Nav1.5 sodium channels in endosomes provide a route for Na+ ion efflux that offsets proton influx during endosomal acidification, a late stage of phagocytosis in macrophages, and demonstrated a role of Nav1.6 sodium channels in controlling macrophage motility via regulation of podosome formation. Microglial migration in vitro is also significantly attenuated by exposure to TTX or phenytoin (Fig. 8.5; Black et al. 2009). Similarly, there is evidence for a role of Nav1.6 in microglial migration (Black et al. 2009).
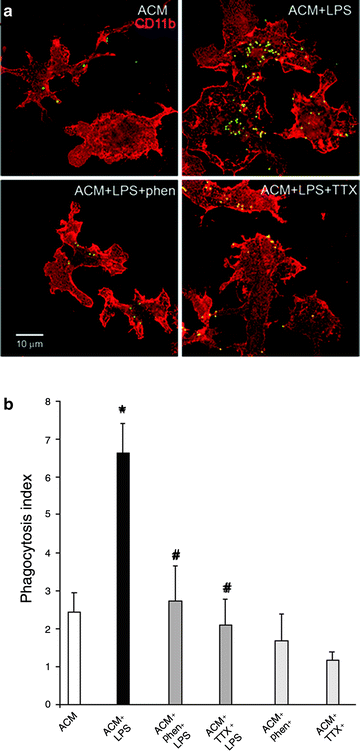
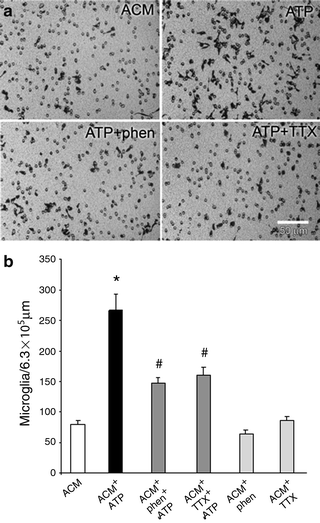
Fig. 8.4
Sodium channel blocking agents inhibit microglial phagocytosis. (a) In astrocyte-conditioned medium (ACM), few latex beads (green) are phagocytosed by CD11b+ microglia (red); in contrast, LPS-activated microglia exhibit increased numbers of phagocytosed latex beads (colocalized latex beads and microglia are yellow). Both phenytoin and TTX attenuate latex bead phagocytosis by LPS-activated microglia. (b) Quantification of phagocytic activity demonstrates a nearly threefold increase in latex bead phagocytosis with LPS activation compared to microglia maintained in ACM only. Phagocytic activity is significantly reduced with phenytoin and TTX treatment in LPS-activated microglia (modified and reprinted with permission from Black et al. 2009)
Fig. 8.5
Sodium channel blockade attenuates microglial migration. (a) Few microglia migrate through the trans-well-membrane pores in ACM only; in contrast, ATP activates a large number of microglia to migrate. Both phenytoin and TTX reduce the number of migrating ATP-activated microglia. (b) Quantification of microglial migration demonstrates a nearly fourfold increase in migrating cells following ATP activation compared to nonactivated microglia. Both phenytoin and TTX (0.3 μM) decrease the number of ATP-activated migrating microglia by ∼50% (modified and reprinted with permission from Black et al. 2009)
Craner et al. (2005) demonstrated that treatment with phenytoin reduces the inflammatory infiltrate with the CNS by 75%. Black et al. (2007) more recently carried out additional studies on the effect of treatment with sodium channel blocking agents on immune cell infiltration and/or functions in EAE. Spinal cord sections of control C57/Bl6 mice labeled for CD45 (leukocyte common antigen), which provides a marker for most immune cells (Sedgwick et al. 1991), exhibited relatively few CD45+ cells (Fig. 8.6). In contrast, untreated mice with EAE mice at 28 days post-MOG injection displayed substantial infiltration of CD45+ immune cells in dorsal, ventral, and lateral funiculi (Fig. 8.6). Mice with EAE that were administered phenytoin (Fig. 8.6) or carbamazepine, however, showed substantially reduced numbers of CD45+ cells in their spinal cords (Black et al. 2007).
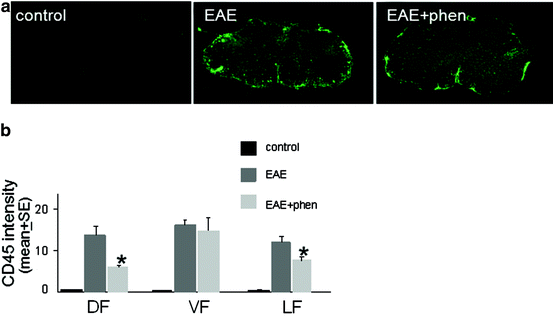
Fig. 8.6
Phenytoin attenuates immune cell infiltration in C57/Bl6 mice with EAE. (a) Low magnification images of cross-sections through lumbar spinal cords labeled for CD45 are shown for control, EAE 35 days postinjection (35 day EAE), and EAE + phenytoin 35 days post-injection (35 day EAE + phen) mice. There is substantial infiltration of CD45+ immune cells in spinal cords of EAE; phenytoin treatment attenuates the infiltration of immune cells. (b) Quantification of CD45+ intensity in dorsal (DF), ventral (VF) and lateral (LF) funiculi of control, EAE, and EAE + phen mice at 35 days post-injection. Phenytoin treatment significantly (*p < 0.05) reduces CD45 intensity in DF and LF in EAE+phen mice compared to untreated EAE mice (modified and reprinted with permission from Black et al. 2007)
Sodium channel blockade with flecainide also reduced spinal inflammation significantly in DA rats with EAE (Bechtold 2004). Quantification of ED-1 immunoreactivity (marking activated macrophages/microglia) in the spinal cords of the rats revealed that flecainide therapy effected a significant reduction in this measure of inflammation during the relapse stage of EAE (flecainide: 6.8% ± 3.0 of spinal cord area; vehicle: 11.5% ± 4.8; p < 0.05). The expression of the inducible form of nitric oxide synthase during the relapse period of EAE was also significantly reduced in the rats treated with flecainide (Fig 8.7; 0.4% ± 1.0), when compared with vehicle-treated animals (2.0% ± 2.2, p < 0.05). In this context, it is interesting that lamotrigine also reduced lymph node cell proliferation in response to phytohemagglutinin (Makowska et al. 2004). Together with the results obtained in mice, these observations suggest that immunomodulation of immune cell functions by sodium channel blocking agents may contribute to the improvement of the clinical course of EAE by these agents.
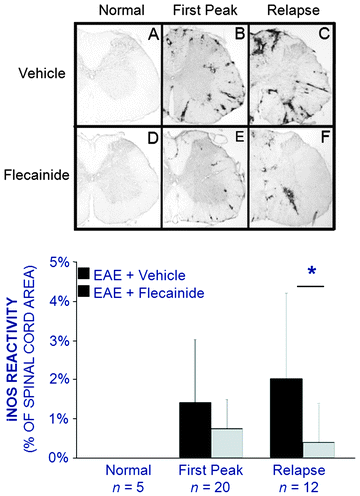
Fig. 8.7
Expression of iNOS in the spinal cords of rats with CR-EAE. iNOS+ cells were not observed in the spinal cords of normal DA rats (A) but were clearly visible during the first peak of CR-EAE (B). The number of iNOS+ cells in the spinal cord increased during the relapse phase of the disease (C). iNOS was not observed in normal rats treated with flecainide (D), but it was observed in the spinal cords of flecainide-treated CR-EAE rats during both the first peak (E) and the relapse (F). However, the magnitude of iNOS reactivity in the rats treated with flecainide was significantly reduced in comparison with rats treated with vehicle (DA Bechtold, P Hassoon and KJ Smith, unpublished observations—modified from Bechtold 2004)
8.3 Withdrawal of Sodium Channel Blockers Can Exacerbate EAE
8.3.1 Neurological Status
The studies described above demonstrated that continued administration of sodium channel blocking agents to mice and rats with EAE can significantly improve the course of the disease; however, whether these protective effects persist following withdrawal of treatment was not known. To examine this question, Black et al. (2007) treated mice with EAE with phenytoin or carbamazepine commencing on day 10 post-MOG inoculation and then withdrew treatment on day 28 (Black et al. 2007). As demonstrated in Fig. 8.8, withdrawal of phenytoin was accompanied by a rapid (within 2 days) exacerbation of the clinical status of mice with EAE; EAE mice with continuous phenytoin treatment did not exhibit exacerbation. The acute withdrawal of phenytoin was associated with a substantial death rate; 59% of the mice died within 11 days following the withdrawal, with most of the deaths occurring within 48 h of the cessation of treatment (Fig. 8.8a). Like withdrawal of phenytoin from mice with EAE, abrupt cessation of carbamazepine treatment was accompanied by acute worsening of the clinical symptoms of EAE, although the death rate following withdrawal was substantially less (Fig. 8.8b; 7.7%; Black et al. 2007). For EAE mice that survived the exacerbation following sodium channel blocker withdrawal, the clinical scores worsened substantially and rapidly approached those of untreated mice with EAE at similar time points. Notably, there were no ill effects observed following withdrawal of phenytoin or carbamazepine from wild-type (non-EAE) mice. Given the findings in EAE, it is interesting that patients with MS can be weaned from sodium channel blocking agents with no major (or perhaps any) detectable worsening of symptoms (see below).
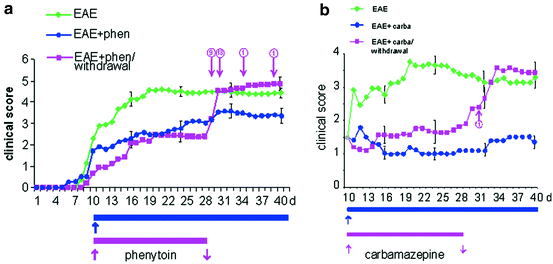
Fig. 8.8
Withdrawal of sodium channel blocking agents exacerbates EAE. (a) Withdrawal of phenytoin increases clinical dysfunction in mice with EAE. Mean clinical scores are shown for C57/Bl6 mice with untreated EAE (green diamonds), EAE + phenytoin (blue circles) and EAE + phenytoin/withdrawal (magenta squares). Phenytoin treatment is indicated by blue (continuous treatment) and magenta (withdrawal) bars. EAE + phenytoin mice exhibit significantly lower clinical scores than untreated mice on all days after day 12 (asterisks omitted for clarity). Withdrawal of phenytoin at day 28 results in rapid worsening of clinical scores. Numbers above magenta arrows indicate number of deaths at each post-withdrawal timepoint. No deaths occurred in EAE and EAE + phenytoin groups of mice from day 28 to 40. (b) Mean clinical scores are shown for C57/Bl6 mice with untreated EAE (green diamonds), EAE + carba (blue circles), and EAE + carbamazepine/withdrawal (magenta squares) commencing on day 10. Carbamazepine treatment is indicated by blue (continuous treatment) and magenta (withdrawal) bars. EAE + carbamazepine mice (blue circles) exhibit significantly lower clinical scores than untreated mice on all days after day 12. Withdrawal of carbamazepine at day 28 results in acute worsening of clinical scores. Number above magenta arrow indicates number of deaths at post-withdrawal timepoint. No deaths occurred in EAE and EAE + carbamazepine groups of mice from day 28 to 40 (modified and reprinted with permission from Black et al. 2007)
8.3.2 Inflammatory Rebound
What is/are the underlying mechanism(s) responsible for exacerbation of EAE following abrupt withdrawal of sodium channel blockers? Since loss of axons has been correlated with accumulation of nonremitting disability in rodent EAE (Wujek et al. 2002), it was conceivable that the observed worsening in EAE following phenytoin or carbamazepine withdrawal was due to massive injury to axons. Quantitative studies, however, demonstrated that there was not a significant difference in the number of axons within dorsal columns of mice with EAE maintained on phenytoin and those in which phenytoin was withdrawn 7–10 days prior (Black et al. 2007). While it is possible that axonal injury had occurred but had not progressed to a quantifiable loss of axons, because Wallerian degeneration of CNS axons can take weeks to become manifest (McDonald 1972), the histological results did not provide evidence for a massive loss of axons.
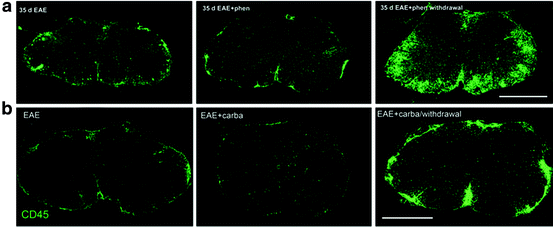
To determine whether inflammation could have contributed to worsening following sodium channel blocker withdrawal, Black et al. (2007) next directed attention to the extent of immune infiltrate into the spinal cords of mice with EAE following phenytoin and carbamazepine withdrawal (Fig. 8.9). As described above, CD45+ cells, which were generally not observed in control mice, were clearly present in the dorsal, lateral, and ventral funiculi of the spinal cord in untreated mice with EAE at 35 days postinjection. When assessed during treatment with phenytoin (Fig. 8.9a) or carbamazepine (Fig. 8.9b) at day 28 postinoculation for EAE, the inflammatory infiltrate was suppressed. Surprisingly, EAE mice treated with phenytoin or carbamazepine from day 10 to day 28 followed by withdrawal at day 28 exhibited a prominent infiltration of inflammatory cells in all funiculi of the spinal cord (Fig. 8.9a, b; Black et al. 2007). Immunocytochemical analysis showed increased numbers of mature T lymphocytes labeled with anti-CD3, helper-inducer T lymphocytes labeled with anti-CD4, cytotoxic/suppressor T lymphocytes labeled with anti-CD8, and activated microglia/macrophages labeled with anti-CD11b and CD68 in the inflammatory infiltrate after phenytoin withdrawal (Black et al. 2007). Additional studies with flow cytometry demonstrated quantitative differences in the inflammatory infiltrate between untreated EAE mice at day 35 postinoculation and EAE mice in which phenytoin was withdrawn at day 28, prior to study at day 35 (Black et al. 2007). There was a 2.5-fold increase in the number of CD3+ cells (T lymphocytes) and a 3.5-fold increase in the number of CD11b+ cells(microglia/macrophages) from the spinal cords and brains of EAE mice following phenytoin withdrawal as compared with untreated EAE mice (Black et al. 2007).
 Immune Modulation and Repair Following Neural Stem Cell Transplantation
Immune Modulation and Repair Following Neural Stem Cell Transplantation
 Effects of Current Medical Therapies on Reparative and Neuroprotective Functions in Multiple Sclerosis
Effects of Current Medical Therapies on Reparative and Neuroprotective Functions in Multiple Sclerosis
 Development of Oligodendrocytes in the Vertebrate CNS
Development of Oligodendrocytes in the Vertebrate CNS
 A Peripheral Alternative to Central Nervous System Myelin Repair
A Peripheral Alternative to Central Nervous System Myelin Repair
 Imaging of Demyelination and Remyelination in Multiple Sclerosis
Imaging of Demyelination and Remyelination in Multiple Sclerosis
 Exogenous Cell Myelin Repair and Neuroprotection in Multiple Sclerosis
Exogenous Cell Myelin Repair and Neuroprotection in Multiple Sclerosis




Related posts:
Immune Modulation and Repair Following Neural Stem Cell Transplantation
Effects of Current Medical Therapies on Reparative and Neuroprotective Functions in Multiple Sclerosis
Development of Oligodendrocytes in the Vertebrate CNS
A Peripheral Alternative to Central Nervous System Myelin Repair
Imaging of Demyelination and Remyelination in Multiple Sclerosis
Exogenous Cell Myelin Repair and Neuroprotection in Multiple Sclerosis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
