Indirect effects
Active (consequences of molecular changes in immune cells exposed to systemic therapy)
Passive (deletion of immune cells or blocking CNS access)
Molecules expressed by modulated immune cells could mediate injury or promote protection/repair of OLs/neurons
Effects of modulated immune cells on CNS microenvironment (astrocytes and microglia) with consequences for OLs/neurons
Loss of molecular mechanisms involved in clearance of tissue injury (e.g., macrophages)
Loss of physiological signals involved in repair and protection (neurotrophins)
Direct effects
Direct interactions with OLs/neurons
Impact on CNS microenvironment (microglia and astrocytes) with consequences for OLs/neurons
9.1.2 Assessment of Effects of Multiple Sclerosis-Directed Immunomodulatory Therapies on Neuroprotection and Repair
Given the complex pathologies of MS, the effects of current and emerging therapies for MS on reparative and neuroprotective functions are likely to reflect their impact on the injury and recovery aspects of the disease. The impact on these aspects need not be congruent as these two phases of the disease may be differentially affected by a particular drug. The emphasis of this chapter will be on agents that are primarily designed to act on the immune aspects of the disease process, as only these are currently approved or in advanced clinical trials. Although considerable data exist regarding the potential benefits of therapies that would be considered to impact directly on neuroprotection and/or repair in animal models of MS, particularly experimental autoimmune encephalomyelitis (EAE), their translation into the clinical MS field has not yet occurred. Examples of these include trophic factors and hormones such as prolactin, estrogens, and progesterone. One notes that at least some of these “neurobiological” directed therapies such as the neurohormones are already known to have effects on the immune system that may or may not be of benefit for MS (Luger et al. 1996).
Table 9.2
Indirect and direct effects of clinically applied therapies on neuroprotection and repair
Indirect effects | Direct effects | |||
|---|---|---|---|---|
Active | Passive | OLs/neurons | CNS microenvironment | |
IFNβ | –Immune deviation (altered cytokine profile) | |||
–Induction of death receptors on immune cells (fas, TRAIL) | –Reduced immune cell trafficking to CNS | |||
–Induction of neurotrophic and growth factors in immune astrocytic, and endothelial cells | –Removal of endogenously produced IFNβ by IFNβ-directed antibodies | |||
GA | –Immune deviation (Th2 bias) that alter microglia antigen presentation | |||
–Induction of neurotrophic factors | ||||
–Increased “protective immunity” | ||||
Antibodies-immune cell and adhesion molecule reactive | –Immune deviation (i.e., reduction in immune cell trafficking across BBB) | –Reduction of immune mechanisms involved in tissue remodeling and regeneration | –Germline antibodies promote remyelination (i.e., IgM) | |
Chemotherapeutic agents | –Reduction of immune mechanisms involved in tissue remodeling and regeneration | –Direct cytotoxicity of OLs/neurons | ||
Myelin vaccines | –Immune deviation | |||
Minocycline | –Impairment of microglia/macrophage function | |||
Statins | –Immune deviation | –Reduced immune cell trafficking to CNS | –Modulation of cytoskeletal and survival properties of progenitors, mature OLs, and neurons | –Inhibition of proinflammatory mediator production by microglia and astrocytes |
–Experimental data indicate effects are dose-and treatment duration-dependent | –Inhibition of astrocyte-mediated glutamate sequestration | |||
FTY 720 (fingolimod) | –Reduction of entry of naive and central memory T cells and B cells into CNS | –Modulation of cytoskeletal and survival properties of progenitors and mature OLs | ||
–Experimental data indicate effects are dose-and treatment duration-dependent | ||||
IFNγ | –Dose-dependent ER stress response confers protection | |||
–Dose-dependent enhancement of progenitor differentiation and remyelination | ||||
The modern therapeutic era of multiple sclerosis began in the early 1990s with the completion of double-blind clinical trials using interferon β (IFNβ) and glatiramer acetate (GA) (Bornstein et al. 1987; Jacobs et al. 1995). These showed partial but sufficiently significant reductions in frequency and severity of disease relapses which resulted in regulatory approval of these agents. The clinical effects of these agents were associated with even more significant decreases in the number of new inflammatory lesions in the CNS, as measured by magnetic resonance imaging (MRI) techniques. MRI studies with GA further documented that the extent of initial injury, as judged by the irreversibility of initial lesions (formation of persistent “black holes”), was reduced by the therapy. Magnetic resonance (MR) spectroscopy studies indicate that IFNβ therapy can result in an increase in N-acetyl aspartate (NAA) signal, a measure of axonal integrity (Narayanan et al. 2001).
Natural history studies from the pretherapeutic era indicated that the frequency of relapses early in the disease course was linked with subsequent development of disability. The relatively short duration (2 years) of the blinded phase of the pivotal clinical trials makes it difficult to conclude whether there was an effect on disability that would reflect mechanisms other than reduced initial tissue injury, such as enhanced repair and delayed neural degeneration. The major clinical and imaging challenge of how to evaluate the effects of therapies on each of these disease aspects (injury, repair, and neuroprotection) is discussed in a separate chapter. An ongoing challenge in both MS and the animal model EAE is to define the extent to which capacity for repair is linked to sites and extent of the initial injury. There is strong documentation that later disease progression can occur in MS in the absence of measurable continued clinical relapses or MRI-defined new lesion formation (Phadke and Best 1983; Gronseth and Ashman 2000; Mews et al. 1998). Long-term follow-up studies, although limited by methodologic problems, suggest that early and prolonged use of the currently approved agents IFNβ and GA in patients with the relapsing form of MS reduces long-term disease progression (Kappos et al. 2006a, b; Miller et al. 2008). However, clinical trials with these immunomodulatory agents in established progressive phases of MS, either primary or secondary progressive, did not lead to their approval for these phases of disease. This is likely consequent to the marked decrease in inflammation within lesions in these phases compared to earlier stages.
9.1.3 Indirect and Direct Effects of Immunomodulatory Therapies on Neural Cells
Interferon beta (IFNβ) and glatiramer acetate (GA) are currently applied therapies which are systemically administered yet not recognized to cross the BBB, indicating that their primary effects are on the systemic immune system and/or on the BBB. Since neither agent completely abrogates continued immune cell trafficking across the BBB, they may still impact on neuroprotection and repair processes within the CNS by modulating the properties of systemic immune cells that may access the brain parenchyma. Such modulation would involve induction of secreted or cell surface molecules that could promote or impede processes of CNS tissue injury and repair. We will refer to these as active indirect mechanisms of action on neuroprotective or repair mechanisms (Table 9.1); specific mechanisms that apply to each of the agents are discussed later in this chapter. Active indirect mechanisms would also apply to evolving immune-directed therapeutic strategies aimed at selectively modulating the properties of disease-relevant immune components, particularly proinflammatory autoreactive T cells. Examples of the “immune deviation” approach include immunizing with native or altered myelin components or their encoding DNAs (Bar-Or et al. 2007; Bielekova et al. 2000) (Table 9.2).
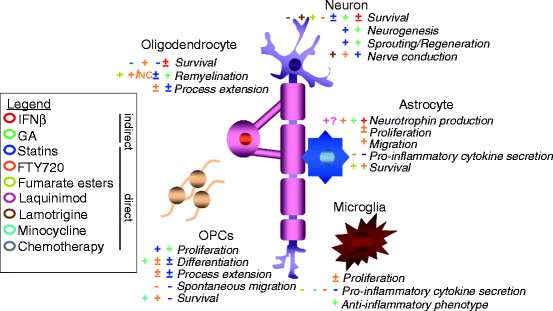
Fig. 9.1
Summary of the effects of MS-directed therapies on neural cell properties important for repair. Listed are properties or responses of neural cells including oligodendrocytes, oligodendrocyte progenitor cells (OPCs), neurons, astrocytes, and microglia. Included are effects on neural cells that may be indirect [i.e., from BBB-impermeable interferon (IFN)-β (red) and glatiramer acetate (GA; green)] or direct [i.e., from BBB-permeable statins (blue), FTY720 (orange), fumarate esters (yellow), laquinimod (pink), lamotrigine (brown), minocycline (turquoise), chemotherapy (gray)]. A promoting/beneficial effect is represented by plus symbol, a negative/detrimental effect is represented by minus symbol, a situation where both positive and negative effects have been observed is represented by plus or minus symbol, no effect/no change is represented by (NC), and where an effect is speculated is represented by question mark
The ongoing development of therapeutic agents in MS has continued to focus on agents that act on the systemic immune system or its capacity to access the CNS. A large family of such agents is comprised of monoclonal antibodies (mabs). Natalizumab, which recognizes the adhesion molecule very late antigen 4 (VLA-4) (also known as α4β1-integrin), markedly reduces relapse frequency and MRI activity (Miller et al. 2003).VLA-4 expression on the surface of endothelial cells is required for binding of activated T lymphocytes to the BBB and subsequent transmigration into the CNS. The blockade of this interaction by natalizumab prevents T cell infiltration into the brain parenchyma of treated MS patients and is thought to underlie its anti-inflammatory properties (Keszthelyi et al. 1996; Yednock et al. 1992). Despite its effects on reducing new lesion formation, clinical trials attempting to show that the therapy-enhanced rate of recovery from relapses were inconclusive (O’Connor et al. 2004). Additional VLA-4 antagonists/inhibitors, such as Firategrast and TV-1102, are being evaluated as potential therapies for MS (Hohlfeld et al. 2011). More recently evaluated monoclonal antibody therapies include daclizumab which blocks the interleukin (IL)-2 receptor (Rose et al. 2007), rituximab and ofatumumab which recognize cluster designation (CD) 20 on B cell lineage cells (Hauser et al. 2008; Buttmann 2010), and alemtuzumab that recognizes CD52 expressed by most lymphoid cells (Coles et al. 2008). Other agents include an oral therapy cladribine, the purine analog which disrupts DNA synthesis and repair in lymphocytes (Giovannoni et al. 2010), and the immunomodulators teriflunomide and ibudilast which inhibit proliferation and activation of lymphocytes (Barkhof et al. 2010; Claussen and Korn 2012). The high degree of efficacy of some of these agents in reducing immune activity or eliminating immune cell trafficking into the CNS raises the issue of whether there could be interruption of physiological immune function within the CNS. We refer to these as passive indirect mechanisms. The balance between potential benefit and toxicity of these agents will determine whether they receive regulatory approval and their use in clinical practice (Tables 9.1, 9.2).
The physiological effects of the immune system are usually considered in context of controlling infection or tumor formation; in this chapter, we focus on the role of the immune constituents in neuroprotection/repair. Inflammation has been shown to be important for the activation of progenitor cells in repair in animal models of demyelination with regard to inducing an activated progenitor phenotype, clearance of inhibitory myelin debris by reactive macrophages, and secretion of molecules that can influence repair (Chari et al. 2006; Glezer et al. 2006; Kotter et al. 2006). Activated immune cells and glia, which can be found in MS lesions, can produce chemokines, cytokines, and growth factors which can promote oligodendrocyte progenitor migration, proliferation, and differentiation and induce remyelination (Arnett et al. 2001; Vela et al. 2002; Maysami et al. 2006; Zhang et al. 2006). Schwartz and colleagues have shown that administration of autoreactive T cells enhances recovery of injured CNS tissue, a process referred to as “protective autoimmunity” (Barouch and Schwartz 2002). MBP-reactive T cells have the potential to be neuroprotective via secretion of BDNF upon activation (Kerschensteiner et al. 1999). In addition, the proinflammatory cytokines IFNγ and tumor necrosis factor alpha (TNFα) are shown to have neuroprotection/repair-related properties in experimental models in which these molecules and/or their receptors have been genetically deleted (Arnett et al. 2001; Rodriguez et al. 2003). Administration of IFNγ in vitro or in vivo in the EAE and cuprizone demyelination models activates an adaptive endoplasmic reticulum (ER) stress response program termed the “integrated stress response” (ISR) that protects mature and oligodendrocyte lineage progenitors against subsequent damage (Lin et al. 2008). However, in small clinical trials, systemic administration of IFNγ exacerbated disease in MS patients, as did anti-TNF antibodies and soluble TNF receptor molecules (Panitch et al. 1987; van Oosten et al. 1996; Gomez-Gallego et al. 2008). Together, these findings suggest that one needs to consider the consequences of therapeutic interventions on the dual functions of immune constituents as promoters/effectors of tissue injury in MS as well as potential contributors to tissue protection/repair (Table 9.2).
The question of the extent of access of systemically administered monoclonal antibodies to the CNS remains open, especially in cases of MS with disrupted BBB. Thus unresolved is the effect that the monoclonal antibody-based therapeutic agents mentioned above may have on adaptive (T cell, B cell) or innate immune activity ongoing within the CNS compartment. The receptors targeted by these monoclonal antibodies are not expressed by primary neural cells. Although immunoglobulin access to the CNS is usually considered as being relatively limited, “therapeutic vaccination” with spinal cord homogenates in experimental CNS closed injury models results in high levels of antimyelin antibodies in the CNS. Experience with CNS amyloid-directed therapies using active immunization regimens and systemic administration of antibody indicate that sufficient antibody can access the CNS to lead to removal of significant amounts of the protein. Sufficient amounts of systemically administered naturally occurring IgM antibodies are able to access the CNS of experimental animals so as to promote remyelination (Bieber et al. 2002).
There are now immune-directed therapies entering clinical use or under investigation in MS that can access the CNS, and thus, one needs to consider their direct effects on neural cells in addition to effects on immune cells within the CNS. Chemotherapeutic agents (cyclophosphamide, mitoxantrone) that have broad cytotoxic effects have been, and continue to be, used for MS patients with worsening disease judged to be unresponsive to the currently approved immunomodulatory agents mentioned previously (Table 9.2). High-dose chemotherapy with agents that can access the CNS is shown by in vivo imaging to be associated with enhanced loss of brain tissue (Freedman 2007). There are newly emerging BBB-permeable therapies that were propelled into MS clinical trials due to observations of their anti-inflammatory properties in experimental systems such as EAE, without extensive data on direct CNS effects. The sphingosine-1-phosphate receptor agonists, including the recently approved FTY720 and the more selective agonist BAF312, presents an example of CNS-accessible agents that act via specific receptors that are broadly expressed within the immune system and the CNS. Lipophilic statins have now been evaluated in clinical trials in relapsing–remitting forms of MS and as a means to prevent development of clinically definite MS in patients with clinical isolated syndromes (CIS) (Vollmer et al. 2004; Menge et al. 2005) (Table 9.2). Some newly emerging oral therapies, including fumarate acid esters (i.e., BG00012), lamotrigine, and laquinimod, have been described as having neuroprotective properties and have been assessed clinically in small-scale exploratory trials. Many of these agents that can access the CNS have primarily been used for non-MS clinical applications, yet their established safety profiles relate to target organs other than the CNS.
9.2 Therapeutic Agents with Indirect Effects on Neural Cells (Fig. 9.1)
9.2.1 Interferon Beta
Interferon beta (IFNβ) (also known as Betaseron, Avonex, and Rebif) is a cytokine that is produced by components of the systemic immune system and also within the CNS by astrocytes (Javed and Reder 2006; Boutros et al. 1997). The finding that IFNβ is most clinically effective in the early, inflammation-driven stages of MS suggests primary effects via immunomodulation. IFNβ induces changes in expression of several hundred genes in immune cells that are relevant for cell activation, migration, proliferation, and effector functions (Javed and Reder 2006). As regards the latter, ligands that are members of the tumor necrosis factor (TNF) superfamily, namely, Fas and TNF-related apoptosis induced ligand (TRAIL), are upregulated by IFNβ (Greil et al. 2003). Both can induce caspase-dependent signaling in target cells that leads to programmed cell death. Fas- and TRAIL-deficient animals develop autoimmune disorders. The extent of Fas and TRAIL upregulation in lymphocytes has been correlated with a favorable disease course in MS (Wandinger et al. 2003). However, the same TRAIL-dependent mechanisms are also shown to underlie lymphocyte-mediated injury of neural cells in slice culture systems (Aktas et al. 2007). IFNβ would have the potential to worsen neural injury by upregulating the expression of TRAIL and Fas ligands on lymphocytes while the receptor counterparts are concurrently upregulated on oligodendroglia under proinflammatory conditions, as observed following exposure to IFNγ in vitro and in MS lesions in vivo (Wosik et al. 2003). Furthermore, upregulation of p53 in human adult CNS-derived OLs in vitro results in increased Fas and TRAIL receptor expression and enhanced susceptibility to ligand-mediated toxicity (Wosik et al. 2003). Increased levels of p53 expression might be expected to occur in response to an array of insults and has been observed in OLs in MS lesions that featured prominent OL loss (Wosik et al. 2003).
Whether IFNβ therapy could enhance repair by sparing neural cells from immune-mediated damage is unclear based on recent animal studies. IFNβ administration to EAE-afflicted animals can dampen or exacerbate clinical symptoms when disease is induced by Th1 versus Th17 lymphocytes, respectively (Axtell et al. 2010). Coadministration with estrogen receptor ligand β decreases EAE severity while preserving axonal densities in the spinal cord (Du et al. 2010), and IFNβ enhances regeneration of axons in a sciatic nerve crush model (Zanon et al. 2010). IFNβ can also induce neurotrophic factors in a number of cell types. Peripheral blood mononuclear cells (MNCs) derived from MS patients are reported to produce neurotrophic factors during IFNβ therapy (Caggiula et al. 2006; Yoshimura et al. 2010). Exposing human brain endothelial cells (HBECs) to MNCs pretreated in vitro with IFNβ, or to MNCs derived from IFNβ-treated patients, induces nerve growth factor (NGF) production by these cells (Biernacki et al. 2005). It has not been determined whether this represents the pro or mature form of the growth factor; the former may have deleterious effects for neural cells (Hempstead and Salzer 2002). IFNβ itself did not induce NGF production by HBECs (Biernacki et al. 2005). The induction of NGF production by HBECs was only observed when MNCs were derived from MS patients at the early onset phase of the disease and correlated inversely with EDSS and MRI lesion burden (Biernacki et al. 2005), suggesting a possible protective rather than injury-mediating function.
In vitro studies have demonstrated that IFNβ can directly influence neural cell properties. NGF is induced in astrocytes following exposure to IFNβ (Boutros et al. 1997). In addition, IFNβ decreases adult mouse neural progenitor apoptosis under death-inducing conditions in vitro (Hirsch et al. 2009) and decreases neurosphere proliferation (Lum et al. 2009). However the systemically administered recombinant cytokine is unlikely to reach high enough levels in the CNS to stimulate this process. IFNβ is also endogenously produced by astrocytes; its activity can be inhibited in vitro by anti-IFNβ antibodies that develop in some IFNβ-treated patients (Shapiro et al. 2006). The titers of the antibodies in serum are sufficiently high in some IFNβ-treated patients such that some access to the CNS would be predicted based on an expected 1:300 ratio between levels of serum and CSF antibodies. Corticosteroid administration at the onset of IFNβ therapy decreases the risk of development of these neutralizing antibodies (Zarkou et al. 2010). Together, this literature suggests that IFNβ therapy has the potential to indirectly influence neural cell properties by modulating peripheral immune components directly or by inducing anti-IFNβ antibodies which interfere with the regulatory function of endogenous IFNβ produced within the CNS. This can potentially have either advantageous or deleterious consequences for neural function or repair.
9.2.2 Glatiramer Acetate
Glatiramer acetate (GA, also known as copaxone or copolymer-1) is a synthetic random polymer of four amino acids (glutamine, lysine, alanine, tyrosine) that are found in the same molar ratio as in MBP. The protective/reparative effects of GA have been linked with the demonstrated capacity of this agent to induce a shift in polarization of GA-reactive T cells from a Th1 to a Th2 phenotype in the periphery which is maintained in the CNS (Aharoni et al. 2000). Th2 polarized lymphocytes can alter the antigen presentation properties of microglia/macrophages, resulting in the subsequent polarization of naïve T cells into a Th2 rather than Th1 phenotype (Kim et al. 2004). CNS injuries have been suggested to demonstrate a shift toward a Th2 cytokine profile. This has been hypothesized to be a necessary component of repair in the CNS subsequent to injury by promoting neural regeneration and preventing autoimmune processes (Hendrix and Nitsch 2007), supporting the concept of “protective immunity” (Kipnis and Schwartz 2002). Th2-polarized lymphocytes can migrate across an endothelial barrier in vitro more effectively than Th1-biased lymphocytes (Biernacki et al. 2001) and may therefore be expected to more readily impact neural cell properties. To be established is whether “immune deviated” myelin antigen-reactive lymphocytes induced by antigen-specific therapies show similar protective/repair properties as GA-reactive T cells.
GA administration to MS patients results in fewer “black holes” on MRI scans, which can be interpreted as less axonal damage or enhanced repair (Bitsch et al. 2001). Systemic administration of GA has been shown to reduce the extent of neuronal injury in a number of experimental models including CNS trauma, neurodegenerative, and autoimmune diseases (Kipnis et al. 2000; Kipnis and Schwartz 2002; Schori et al. 2001; Aharoni et al. 2003; Benner et al. 2004). For instance, GA enhanced OPC numbers and promoted remyelination in a rodent spinal cord demyelination model (Skihar et al. 2009) and prevented demyelination and neuronal loss, stimulated neurogenesis and remyelination, and encouraged axonal sprouting/regeneration in relapse-remitting and chronic EAE (Aharoni et al. 2005, 2008, 2011). The protective/repair effects of GA-reactive T cells within the CNS, whether completely related to their Th2 properties or not, could be mediated directly on the target cell or indirectly via modulation of disease-relevant properties of glial cells. Both in vitro and in vivo studies have demonstrated that GA induces production of neurotrophins such as BDNF, NT3, NT-4 (Aharoni et al. 2003; Ziemssen et al. 2002; Skihar et al. 2009), growth factors such as IGF-1 (Skihar et al. 2009), and cytokines such as TGFβ (Aharoni et al. 2003, 2005). These can be predicted to protect neural cells against insults (Riley et al. 2004), rescue degenerating neurons (Riley et al. 2004), and affect regeneration of mature OLs and their progenitors (Althaus 2004). Administration of GA to EAE-afflicted animals at various clinical stages resulted in a sustained augmentation of levels of neurotrophins in infiltrating lymphocytes, neurons, astrocytes, and newly migrated neural progenitors within the lesion resulting in attenuation of axonal damage (Aharoni et al. 2003). GA also increases growth and neurotrophic factor production in remyelinating lesions of the rodent spinal cord, and by GA-reactive lymphocytes in vitro, resulting in enhanced repair and OPC specification/differentiation, respectively (Aharoni et al. 2008; Skihar et al. 2009; Ziemssen et al. 2002; Zhang et al. 2010).
Other glial cell functions that could be influenced by transmigrated immune cells in GA-treated patients would include the capacity to produce or remove potential injury mediators (such as glutamate and free radicals), production of trophic molecules, and expression of signaling molecules that modulate migration, differentiation, and myelin production by progenitor cells. Some have suggested that GA-carrying dendritic cells have the potential to cross the BBB (Liu et al. 2007), raising the potential of thus far uninvestigated direct effects of GA on neural cells.
9.3 Therapeutic Agents with Direct Effects on Neural Cells
9.3.1 Chemotherapeutic Agents
A number of cytotoxic agents have been used typically for patients who fail conventional therapies. Examples of such therapies include cyclophosphamide, mitoxantrone, and cladribine. Subcutaneous administration of cladribine (2-chlorodeoxyadenosine) is associated with decreased levels of proinflammatory cytokines and chemokines in the CSF of treated MS patients in remission, possibly indicating a potential effect on CNS cells in addition to peripheral suppression (Bartosik-Psujek et al. 2004). A strategy being adopted from other autoimmune disorders is to administer such agents as initial induction therapies to be followed by immunomodulatory therapy. Such therapies can induce initial loss of brain volume that cannot be attributed to eliminating inflammation. This was observed in the Canadian immunoablative therapy protocol, in which patients underwent serial MRI studies before and after the immunoablation phase that included administration of CNS-accessible agents (busulfan), followed by infusion of autologous bone marrow-derived stem cells (Freedman 2007).
9.3.2 Minocycline
Minocycline is a semisynthetic tetracycline that has been in clinical use for nonneurologic indications (e.g., acne) that readily crosses the BBB. The agent has been shown to alleviate CNS pathology in a number of neurodegenerative and inflammatory animal models including stroke, spinal cord injury, Parkinson’s disease, and multiple sclerosis (Arvin et al. 2002; Brundula et al. 2002; Popovic et al. 2002; Wu et al. 2002; Lee et al. 2003; Fox et al. 2005; Nikodemova et al. 2007, 2010). Mechanisms held to account for this include direct effects on blockade of microglia/macrophage activation, inhibition of matrix metalloproteinases, and antiapoptotic effects (Yrjanheikki et al. 1998; Popovic et al. 2002; Elewa et al. 2006; Nikodemova et al. 2007), as well as indirect effects by limitation of CD4+/CD8+ T cell entry into the CNS and peripheral (but not central) immune deviation (Popovic et al. 2002; Nikodemova et al. 2010). Minocycline can also promote the survival and myelination of OPCs transplanted into rodents (Zhang et al. 2003). Clinical trials in MS patients have shown minocycline to beneficial in reducing the number of active scans and minimizing brain volume change (Zhang et al. 2008). However, the neuroprotective properties of minocycline are unclear at the moment given some reports of worsened disease or lack of neuroprotection in animal models (Saganova et al. 2008); improved outcome appears to depend on early initiation of treatment, high dosage, and direct delivery to the CNS (Xue et al. 2010). Of concern are recent clinical trial results showing that minocycline had a harmful effect on patients with amyotrophic lateral sclerosis, an effect not predicted by studies in the SOD1 animal model of this disease (Gordon et al. 2007).
9.3.3 Statins
Statins describe a class of drugs that competitively inhibit the rate-limiting enzyme in the mevalonate pathway, 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG CoA) reductase (HMGR), primarily reducing the biosynthesis of cholesterol and the posttranslational lipid moiety attachments, isoprenoids (Edwards and Ericsson 1999). Their beneficial effects in EAE were attributed to their systemic immune-regulatory actions resulting in changes in protein isoprenylation (Greenwood et al. 2006; Peng et al. 2006; Pahan et al. 1997; Youssef et al. 2002). Based on these studies, simvastatin was selected for a clinical trial in MS and was found to reduce the number of newly emerging lesions by MRI at a dose substantially higher than those typically prescribed for hypercholesterolemia (Vollmer et al. 2004). Statins dampen the expression of proinflammatory cytokines, chemokine receptors, adhesion molecules, and MMPs that would normally facilitate lymphocyte transendothelial migration (Lindberg et al. 2005; Pahan et al. 1997). Additionally, lipophilic statins (i.e., lovastatin, simvastatin) can passively diffuse through cell membranes and directly access the CNS (Saheki et al. 1994). The expression of HMGR in all cell types along with the penetration of this therapy into the brain highlights the potential direct effects of statins on neural cell properties. Accordingly, statins have been attributed neuroprotective properties in neurodegenerative models of stroke, vascular dementia, Alzheimer’s disease, and Parkinson’s disease, although the mechanisms through which this occurs are not yet fully understood (Wang et al. 2011).
Statins can prevent the release of proinflammatory mediators such as prostaglandin E2, nitric oxide, TNFα, IL-1β, and IL-6 from activated microglia and astrocytes (Pahan et al. 1997; Tringali et al. 2004). Treatment of astrocytes with lovastatin also impairs the function of glutamate synthetase which normally degrades excessive cytotoxic amounts of glutamate sequestered from the extracellular environment (Chou et al. 2003). In vitro studies have also indicated that statins can have variable effects on neuronal morphology and cell survival depending on dose, cell maturity, neuronal subtype, length of treatment, and environmental stress (Kumano et al. 2000; Michikawa and Yanagisawa 1999). Simvastatin has been shown to promote neuritic outgrowth on myelin substrates that would normally prevent regeneration and growth of axons (Holmberg et al. 2006). Statins also have beneficial effects on nerve conduction and generation of neuroblasts in neurodegenerative models (Tarhzaoui et al. 2009; Chen et al. 2009). In vitro studies have demonstrated the ability of lipophilic statins to influence cellular processes in human and rodent OPCs that have been implicated in the remyelination process (Miron et al. 2007; Paintlia et al. 2005). For instance, simvastatin treatment can induce an initial process extension, enhanced differentiation, and impaired spontaneous migration via inhibition of isoprenoid synthesis (Miron et al. 2007). Prolonged treatment induced process retraction via inhibition of cholesterol production and decreased viability and cell death caused by interference with both isoprenoid- and cholesterol-dependent functions in the cells (Miron et al. 2007). Mature oligodendrocytes treated with statins demonstrate similar responses to OPCs with regard to cytoskeletal dynamics and survival (Miron et al. 2007). Statin treatment may have altered the activity levels of the normally isoprenylated Rho GTPases to regulate effects on process extension, cell survival, differentiation, and migration. In addition, statin exposure may have altered the dynamics of cholesterol-rich lipid rafts important for OL process outgrowth (Decker and Ffrench-Constant 2004; Michikawa and Yanagisawa 1999).
Myelin contains a high content of lipids which consist of the majority of its dry weight, over a quarter of which is composed of cholesterol (Morell and Jurevics 1996). Importantly, brain-derived cholesterol cannot be replaced by dietary or peripheral sources (Jurevics and Morell 1995; Morell and Jurevics 1996; Saher et al. 2005). Short-term daily treatment of EAE-afflicted animals with an immunomodulatory dose of lovastatin was shown to increase myelin recovery in the spinal cord by enhancing survival as well as proliferation and differentiation of OPCs (Paintlia et al. 2004). This was attributed to the creation of an environment favorable to remyelination by altered inflammation and neurotrophic factor production. However, these studies could reflect either a direct impact of the statin on neural cells or a sparing of immune-mediated injury consequent to anti-inflammatory activity. This was addressed by using a relatively non-immune-mediated model of brain-based demyelination (oral cuprizone administration) to assess the direct impact of statins on myelin maintenance and remyelination (Klopfleisch et al. 2008; Miron et al. 2009). Both short-term and long-term administration of an immunomodulatory dose of the more potent simvastatin impaired remyelination when statin was administered during the period of concomitant de- and remyelination, the entire period of OPC responses to demyelination, or the remyelination period alone. Effects were associated with a decrease in OPC numbers when the drug was applied during the period of concomitant de- and remyelination, and an increase in OPCs in an immature state when statin was applied during the remyelination period suggesting a block in differentiation (Miron et al. 2009). Together, these findings suggest the potential of lipophilic statins to have both deleterious and advantageous effects on the function of various neural cell types and repair/regeneration in the CNS.
9.3.4 FTY720
FTY720 (fingolimod/Gilenya) is a recently clinically approved oral agent for the treatment of relapsing MS that induces profound reduction in numbers of lymphocytes circulating in the peripheral blood due to its capacity to enhance ingress and reduce egress of CCR7-expressing naïve and central memory lymphocytes from regional lymph nodes (Compston and Coles 2002). This agent was initially evaluated (and concluded to be ineffective) as a therapy to prevent organ rejection in transplant patients. Based on these immunomodulatory properties, this agent has been successfully used in clinical trials in relapsing–remitting MS and is being evaluated in primary progressive MS clinical trials (Kappos et al. 2006a, b). FTY720 decreases the rate of neurodegeneration in patients compared to an IFNβ-1a-treated cohort, as measured by brain volume loss (Khatri et al. 2011). The clinically administered native form of FTY720 can readily cross the BBB by virtue of its lipophilicity (Sanchez and Hla 2004). Once it has entered the brain parenchyma, it is rapidly phosphorylated by sphingosine kinase (SphK) and consequently biotransformed into an analogue of the endogenous bioactive lysophospholipid, sphingosine-1-phosphate (S1P) (Billich et al. 2003; Zemann et al. 2006). The active phosphorylated form of the drug can bind to four of five known G protein-coupled receptors which belong to the endothelial differentiation gene-related (EDG) family, termed S1P1, 3, 4, and 5 or EDG 1, 3, 6, and 8, respectively (Davis et al. 2005; Mandala et al. 2002). Generally, S1P3/4/5 activation leads to G12/13 signaling and process retraction via the cytoskeletal modulator RhoA GTPase (Jaillard et al. 2005; Toman et al. 2004). S1P1 signaling to Gi/o is associated with Rac1, Ras, and extracellular signal-related kinase (ERK) 1/2 activation and subsequent process extension, survival, migration, and proliferation (Goetzl and Rosen 2004; Toman et al. 2004). S1P receptors are ubiquitously expressed throughout the immune and central nervous systems. The biological basis behind its anti-inflammatory effects reflects the ligation of the bound S1P receptor on circulating naïve and central memory lymphocytes, with eventual endocytosis of the bound receptor, downregulation of the receptor at the mRNA level, and subsequent blockade of S1P1-dependent efflux from secondary lymph nodes to target organs such as the brain (Goetzl and Rosen 2004; Liu et al. 1999; Sawicka et al. 2005). Fingolimod has previously been shown to have neuroprotective effects in experimental models of spinal cord injury, stroke, and multiple sclerosis (Zhang et al. 2009; Hasegawa et al. 2010; Foster et al. 2007).
Audoradiographical analysis of rodents that were orally administered C14-labelled FTY720 for 1 week indicate that both the native drug and the phosphorylated metabolite entered the brain parenchyma, steadily increased in concentration over time within this compartment, and was concentrated in myelin sheaths (Foster et al. 2007). Levels of the bioactive form of FTY720 are within the micromolar range in the brain, whereas subnanomolar concentrations are measured in the CSF (Foster et al. 2007).
FTY720 treatment of relapsing EAE animals, either prophylactically or during the chronic phases of the disease, can prevent or reverse clinical disability, respectively (Fujino et al. 2003; Webb et al. 2004; Kataoka et al. 2005; Al-Izki et al. 2011), whereas a recent study has demonstrated that clinical deterioration is not prevented when FTY720 is administered during progressive EAE (Al-Izki et al. 2011). Microarray profiling has revealed that FTY720 treatment of EAE animals also results in a downregulation of proinflammatory cytokine and chemokine-related genes and an increase in transcript levels of myelin-related genes (Foster et al. 2007). Given that the clinically administered native form of FTY720 can readily cross the BBB, such responses in the EAE model could reflect effects in the periphery, at the BBB, and/or on neural cells. The efficacy of FTY720 in relieving clinical disability in the later stages of EAE where inflammation has subsided suggests a neuroprotective effect (Fujino et al. 2003; Webb et al. 2004; Foster et al. 2007).
The combination of FTY720’s crossing of the BBB and the expression of S1P1 and S1P3 on human-derived endothelial cells indicates the potential of this agent to impact on endothelial barrier properties in treated patients (Lin et al. 2007). FTY720 has been shown in vivo to promote adherens junction assembly between endothelial cells and decrease BBB permeability via S1P1 signaling (Lin et al. 2007; Foster et al. 2009). Astrocytes and microglia also have the potential to respond to S1P receptor signaling, with the expression of S1P1, 3, and 5 receptor isoforms. FTY720 treatment of astrocytes in vitro promotes their migration and survival via S1P1 signaling (Mullershausen et al. 2007; Osinde et al. 2007). Selective loss of S1P1 expression in astrocytes abrogates the protective effect of FTY720 in EAE (Choi et al. 2011). These findings suggest that S1P receptor signaling on astrocytes can influence cellular events that contribute to the inflammatory response and/or to a nonsupportive microenvironment, i.e., a parallel with the glial scar found in MS lesions. Studies using rat-derived microglia have indicated that their relative S1P receptor levels are modulated based on their activation state and that S1P receptor engagement on these cells alters their cytokine profile (Tham et al. 2003). This agent also increases microglia and astrocyte cell numbers ex vivo and in vivo during remyelination (Miron et al. 2010; Kim et al. 2011). Furthermore, FTY720 inhibits macrophage migration into inflammatory lesions (Zhang et al. 2007). Together these data indicate that FTY720 may influence their roles in phagocytosing debris within lesions and regulating the immune responses within the CNS. FTY720 also normalizes electrophysiological responses in EAE-afflicted animals (Balatoni et al. 2007). Although FTY720 has been shown to induce apoptosis in rat neurons, this only occurs at a concentration which would be supraphysiological in the CNS (Oyama et al. 1998). Although FTY720 does enter the brain parenchyma, it is not primarily localized to neurons suggesting limited potential to impact neuronal properties (Foster et al. 2007).
Rodent-derived mature OLs and OPCs express S1P receptors both in vitro and in vivo, in the relative abundance of S1P5 > S1P1 = S1P2 > S1P3 for OLs and S1P1 = S1P2 = S1P5 > S1P3 for OPCs (Yu et al. 2004; Jaillard et al. 2005; Novgorodov et al. 2007; Miron et al. 2007, 2008b). FTY720 has been shown to induce transient S1P3/5-dependent process retraction with subsequent S1P1-dependent process extension in human OPCs and adult mature OLs but only in rodent OLs matured in vitro when coapplied with neurotrophic factors (Miron et al. 2007, 2008a, b; Coelho et al. 2007). FTY720 induced cyclic down- and upregulation of S1P1 and S1P5 mRNA transcripts over time in cultured oligodendroglial cells, in contrast to the sustained downregulation of S1P1 observed in lymphocytes exposed to the drug in vivo (Miron et al. 2007, 2008a, b). FTY720 inhibits OPC differentiation and spontaneous migration yet does not influence directed migration to the chemoattractant PDGF (Jung et al. 2007). FTY720 was able to enhance OPC and OL survival under death-inducing conditions via S1P1 and S1P5 signaling, respectively (Miron et al. 2007, 2008a, b; Coelho et al. 2007; Jung et al. 2007). Contrasting reports exist on the ability of oral administration of FTY720 to promote OL survival and myelin integrity in experimental models of de- and remyelination (Kim et al. 2011; Hu et al. 2011). Kim et al. reported that 1 mg/kg/day FTY720 orally administered during 6 weeks of cuprizone-induced demyelination attenuated injury to OLs, neurons, and myelin, causing an increase in the percentage of myelinated fibers compared to control and associated with a reduced astro- and microgliotic response. However, when FTY720 was administered during the entire period of OPC responses (6 weeks during cuprizone administration and return to normal diet), an increase in astrogliosis was observed, thereby indicating that FTY720 can induce differential neural responses depending on the time it is administered following injury. Accordingly, Mi et al. observed that FTY720 (1 mg/kg/day) orally administered for 2 weeks at 4 weeks after the onset of cuprizone diet did not have any protective effect on the proportion of myelinated axons or lesion volume (Hu et al. 2011). Neither group observed an increase in remyelination in vivo, perhaps reflecting the difficulty in assessing enhanced remyelination in young animals where this process is rapid and effective. In contrast, application of physiological doses of fingolimod to organotypic mouse cerebellar slice cultures, which maintain the physiological cell–cell interactions and have mature compact myelin, enhanced remyelination following toxin-induced demyelination via S1P3/5 signaling (Miron et al. 2010). This was associated with process extension in both OPCs and mature OLs, as well as an increase in microglial and astrocytic components, which may have contributed to the repair process by production of factors beneficial to OPC and OL function. Differences with in vivo observations may indicate limited bioavailability of the drug in an animal compared to direct application in vitro. Mi et al. (2011) showed that FTY720 caused a decrease in developmental myelination in slices of P17 rat corpus callosum, which may reflect either differential susceptibility of myelin and OLs to FTY720 under myelinating and remyelinating conditions, or distinct responses of specific white matter tracts to the drug. Overall, the net effect of FTY720 on neural cells will reflect the relative levels of the S1P receptor subtypes on cells of interest, the modulation of these receptor levels in response to the environment, and counteracting signals emanating from the inflamed brain. Efforts are underway to assess more selective S1PR agonists, such as BAF312, which specifically binds S1P1 and S1P5, shows encouraging results in decreasing disease severity in EAE (Nuesslein-Hildesheim et al. 2010).
Related posts:
 Immune Modulation and Repair Following Neural Stem Cell Transplantation
Immune Modulation and Repair Following Neural Stem Cell Transplantation
 Development of Oligodendrocytes in the Vertebrate CNS
Development of Oligodendrocytes in the Vertebrate CNS
 A Peripheral Alternative to Central Nervous System Myelin Repair
A Peripheral Alternative to Central Nervous System Myelin Repair
 Endogenous Remyelination in the CNS
Endogenous Remyelination in the CNS
 Exogenous Cell Myelin Repair and Neuroprotection in Multiple Sclerosis
Exogenous Cell Myelin Repair and Neuroprotection in Multiple Sclerosis
 Axonal Protection with Sodium Channel Blocking Agents in Models of Multiple Sclerosis
Axonal Protection with Sodium Channel Blocking Agents in Models of Multiple Sclerosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


