Fig. 4.1
(a) Electron micrograph of myelinated axons and axons that have been remyelinated following ethidium bromide injection into the adult rat caudal cerebellar peduncle. The myelinated axons have clearly discernible myelin sheaths whose thickness is proportional to the axon diameters (compare M1 with M2). The remyelinated axons are also clearly discernible on account of the relatively thin myelin sheaths, whose thinness is consistent and independent of the axon diameter (compare R1 with R2). Thus, remyelination is easy to identify for larger diameter axons; however, for small diameter axons, the distinction between myelinated and remyelinated becomes difficult (note the small diameter axons in this image—are they myelinated or remyelinated?) (b) The relationship between the axon diameter (x) and the myelinated axon (y) is expressed as the g ratio: the thinner the myelin sheath, the higher the g ratio, and hence remyelinated axons, unless very small diameter, have g ratios that are higher than myelinated axons. (c) In developmental myelination, as the axon diameter increases, the myelin sheath thickness increases (see M1 with M2 in a), whereas in remyelination, the myelin sheath thickness remains the same regardless of the diameter (see R1 with R2 in a). Thus, remyelination in large diameter axons is easy to distinguish from myelination—but as the axon diameter decreases, this distinction becomes more difficult such that for the smallest diameter axons it can be all but impossible (see Stidworthy et al. 2003)
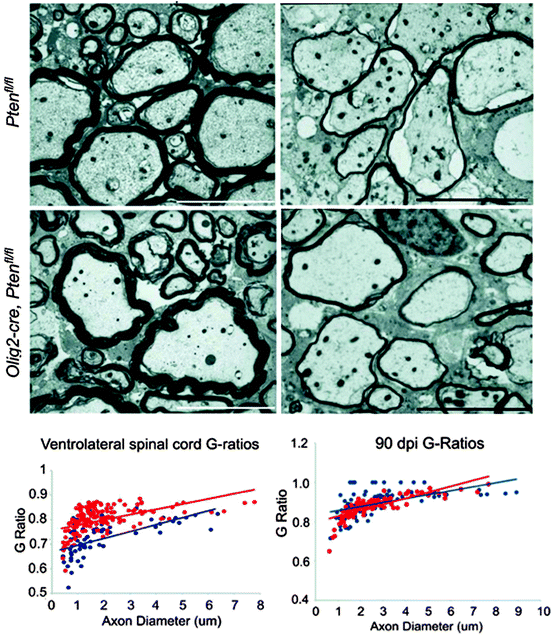
Fig. 4.2
When the Akt pathway is over-activated in mice where Pten has been conditionally deleted from oligodendrocyte lineage cells, the myelin sheath thickness is increased in relationship to axon diameter (i.e. the g ratio decreases). This is shown in the two electron micrographs on the left taken from the ventrolateral spinal cord white matter of adult control mice (Ptenfl/fl) and mice in which Pten is deleted from the oligodendrocyte lineage (Olig2-Cre, Ptenfl/fl). These images are confirmed below in the g-ratio plots (blue dots = Ptenfl/fl, red dots = Olig2-Cre, Ptenfl/fl). Following lysolecithin-induced demyelination on the ventrolateral white matter, the remyelinated axons in both experimental groups are the same (electron micrographs and g-ratio plots on right) (from Harrington et al. 2010)
4.3 Remyelination Is the Normal Response to Demyelination
Remyelination can be viewed as a regenerative process sharing many common features with regenerative processes occurring in other tissues of the body and as being the default response to demyelination. This viewpoint is based on evidence from both experimentally-induced and clinical demyelination. When demyelination is induced by toxins injurious to oligodendrocytes and myelin (e.g. dietary cuprizone or direct delivery of lysolecithin or ethidium bromide), then remyelination usually proceeds to completion, albeit in an age-dependent manner (Blakemore and Franklin 2008). Similarly, there is evidence that axons undergoing primary demyelination in experimental or clinical traumatic injury undergo complete remyelination and that the persistence of chronically demyelinated axons is unusual (Lasiene et al. 2008). An exception is when demyelination is induced by or associated with the adaptive immune response, such occurs in the autoimmune-mediated condition multiple sclerosis (MS) and in its laboratory animal model experimental autoimmune encephalomyelitis (EAE). In this circumstance, remyelination is required to take place in an environment intrinsically hostile to the oligodendrocyte lineage. Thus, remyelination failure associated with MS (and EAE) can be seen not as a generic feature of remyelination biology but rather as a feature of specific disease states. However, even in MS, a disease normally associated with failed or inadequate remyelination, there is evidence that in some patients, complete remyelination occurs in a significant proportion of lesions (Patrikios et al. 2006; Patani et al. 2007; Goldschmidt et al. 2009; Piaton et al. 2009). Similarly, remyelination can be extensive in EAE, and models with significant persistent demyelination are unusual (Hampton et al. 2008; Linington et al. 1992). Remyelination appears to be especially efficient following demyelination of cerebral cortical grey matter in both experimental models (Merkler et al. 2006) and clinical disease (Albert et al. 2007), although the reason for this is unclear.
4.4 Remyelination Restores Function and Protects Axons
Remyelination restores saltatory conduction and reverses functional deficits (Liebetanz and Merkler 2006; Jeffery et al. 1999; Smith et al. 1979). Compelling evidence in support of functional restoration by remyelination has recently been provided by an unusual demyelinating condition in cats where the reversal of clinical signs is associated with spontaneous remyelination (Duncan et al. 2009).
A further and key function of remyelination is axon survival (Irvine and Blakemore 2008). Axonal and neuronal loss is now recognised as the major cause of chronic progressive disease (Trapp and Nave 2008), and it occurs as a secondary consequence of demyelination in addition to any primary effect of inflammation. Such a hypothesis explains why patients taking immunosuppressive therapies or with apparently quiescent disease still show increasing disability and progression, as these patients will have persistent demyelination as a result of failure of remyelination even in the absence of active disease. It should be noted that remyelination is not required for resolution of symptoms in an acute relapse: this likely results from resolution of inflammation and adaptive responses of the axon to restore conduction.
Evidence that myelin is required for axon survival is based on observations of genetic mouse models and studies of human pathology (Nave and Trapp 2008). Transgenic mice lacking CNP or PLP show long-term axonal degeneration, even in the presence of myelin sheaths that are either ultrastructurally normal or show only minor abnormalities (Griffiths et al. 1998; Lappe-Siefke et al. 2003). Further analysis of the PLP mutant mice has revealed a disturbance in axoplasmic transport in the absence of PLP (Edgar et al. 2004) and has led to the identification of myelin-associated sirtuin 2 as a potential mediator of long-term axonal stability (Werner et al. 2007). Myelin is also important for axon survival in humans, as patients with Pelizaeus–Merzbacher disease (PMD) caused by mutations in PLP show axon loss (Garbern et al. 2002), and studies of MS autopsy tissue show that axon preservation is seen in those areas where remyelination has occurred (Kornek et al. 2000). Axon degeneration has recently been observed as a consequence of genetically induced oligodendrocyte-specific ablation, even in Rag 1-deficient mice that have no functional lymphocytes (Pohl et al. 2011), thus providing compelling evidence that axon survival is dependent on intact oligodendrocytes and that axon degeneration in chronically demyelinated lesions can occur independently of inflammation. These observations imply that remyelination therapies will promote axon sparing in MS by the production of an oligodendrocyte-derived trophic factor signal to the axon.
4.5 The Mechanisms of Remyelination
4.5.1 Oligodendrocyte Precursor Cells Are the Main Source of New Myelin-Forming Oligodendrocytes
Remyelination involves the generation of new mature oligodendrocytes since (1) there is a greater number of oligodendrocytes within an area of remyelination compared to the equivalent area before myelination (Prayoonwiwat and Rodriguez 1993) and (2) remyelination occurs within areas depleted of oligodendrocytes (Sim et al. 2002b). In the vast majority of cases, the new oligodendrocytes that mediate remyelination are derived from a population of adult CNS stem/precursor cells, most often referred to as adult oligodendrocyte precursor (or progenitor) cells and sometimes called NG2 cells (in this chapter, OPC will generally refer to these cells). These multiprocessed proliferating cells are widespread throughout the CNS, occurring in both white matter and grey matter at a density similar to that of microglial cells (5–8 % of the cell population) (Horner et al. 2000; Richardson et al. 2011; Dawson et al. 2003). Adult OPCs are derived from their developmental forebears, and the two cells share many similarities, although the adult cell has a longer basal cell cycle time and slower rate of migration (Wolswijk and Noble 1989). Relevant to remyelination, the adult OPC can be induced to proliferate and migrate like perinatal cells in vitro by the growth factors PDGF and FGF (Wolswijk and Noble 1992), both of which are significantly upregulated during remyelination (Hinks and Franklin 1999).
Evidence obtained using Cre-lox fate mapping in transgenic mice following experimental demyelination has shown that OPCs produce the vast majority of remyelinating oligodendrocytes (Zawadzka et al. 2010; Tripathi et al. 2010) (Fig. 4.3). Remyelinating oligodendrocytes can also come from the stem and precursor cells of the adult subventricular zone (SVZ), either from the precursor cells contributing to the rostral migratory stream (RMS) (Nait-Oumesmar et al. 1999) or from the type B, GFAP-expressing stem cells of the SVZ per se (Menn et al. 2006). However, the contribution that SVZ-derived cells make relative to that of local OPCs may be relatively small, and their contribution to repair away from white matter tracts that are not close to the SVZ is likely to be negligible.
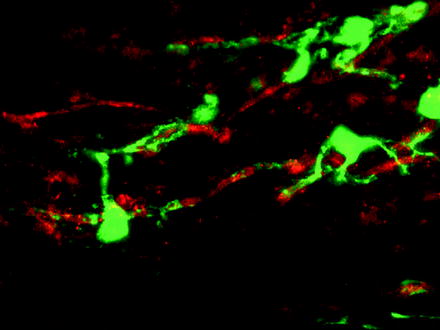
Fig. 4.3
OPCs can be labelled by a Cre-lox labelling strategy enabling the fate of these cells following lysolecithin-induced demyelination to be traced. In this confocal image, a YFP (green)-labelled cell is associated with a PLP+ (red) myelin sheath providing evidence that the labelled OPC has differentiated into a myelin sheath forming oligodendrocyte responsible for remyelination (see Zawadzka et al. 2010)
4.5.2 Remyelination Requires the Activation, Recruitment and Differentiation of Adult Oligodendrocyte Precursor Cells
In response to injury, OPCs in the vicinity undergo a switch from an essentially quiescent state to a regenerative phenotype. This activation is the first step in the remyelination process and involves not only changes in morphology but also upregulation of several genes, many associated with the generation of oligodendrocytes during development such as the transcription factors Olig2, Nkx2.2, MyT1 and Sox2 (Watanabe et al. 2004; Fancy et al. 2004; Shen et al. 2008). The activation of OPCs is likely to be in response to acute injury-induced changes in microglia and astrocytes, two cell types exquisitely sensitive to disturbance in tissue homeostasis (Glezer et al. 2006; Rhodes et al. 2006). These two cell types, themselves activated by injury, are the major source of factors that induce the rapid proliferative response of OPCs to demyelinating injury. This response is modulated by the levels of the cell cycle regulatory proteins p27Kip-1 and Cdk2 (Crockett et al. 2005; Caillava et al. 2011) and is promoted by the growth factors PDGF and FGF (Murtie et al. 2005; Woodruff et al. 2004) and doubtless other factors associated with acute inflammatory lesions and demonstrated to have OPC mitogenic effects in tissue culture (Vela et al. 2002). Semaphorins have emerged as important regulators of OPC migration following demyelination. Developmental studies first identified semaphorins 3A and 3F as repulsive and attractive guidance cues for OPCs, respectively. It has subsequently been shown that adult OPCs express class 3 semaphorin receptors, neuropilins and plexins and that neuropilin expression increases after demyelination. Gain and loss of function experiments have shown that semaphorin 3A impairs OPC recruitment to the demyelinated area, while semaphorin 3F overexpression accelerates not only OPC recruitment but also remyelination rate (Piaton et al. 2011). The population of areas of demyelination by OPCs is referred to as the recruitment phase of remyelination and involves by OPC migration in addition to the ongoing proliferation.
For remyelination to be complete, the recruited OPC must next differentiate into remyelinating oligodendrocytes—the differentiation phase. This phase encompasses three distinct steps—establishing contact with the axon to be remyelinated, expression of myelin genes and generation of myelin membrane and finally wrapping and compacting to form the sheath. Despite these being fundamental properties of oligodendrocytes, we still have an incomplete understanding about how axoglial contact is established and how this interaction then regulates, within each individual cell process, the morphological changes that constitute myelination. That said, some molecules have been shown to contribute to the regulation of differentiation, and it is clear that the differentiation of OPCs into myelinating oligodendrocytes in development and during the regenerative process shares many similarities (Fancy et al. 2011a). FGF plays a key role in inhibiting differentiation as well as promoting recruitment and thereby regulates the correct transition from the recruitment to the differentiation phases (Armstrong et al. 2002), and IGF-I is another factor that plays major roles in both processes (Mason et al. 2003). Recently, it has been shown that semaphorin 3A, in addition to its role in OPC recruitment (Piaton et al. 2011), is also an inhibitor of OPC differentiation (Syed et al. 2011). LINGO-1, a component of the trimolecular Nogo receptor, has been found to be a negative regulator of oligodendrocyte differentiation in development (Mi et al. 2005), while mice deficient in LINGO-1 or treated with an antibody antagonist against LINGO-1 exhibited increased remyelination and functional recovery from experimental autoimmune encephalomyelitis (EAE) (Mi et al. 2007). The canonical Wnt pathway has recently emerged as a very powerful negative regulator of oligodendrocyte differentiation in both development and remyelination (Ye et al. 2009; Fancy et al. 2009). For example, constitutive induction of Wnt signalling in oligodendrocyte lineage cells by using transgenic mice that actively express a dominant active β-catenin gene results in mice displaying severe tremor and ataxia within 1 week after birth due to delayed oligodendrocyte differentiation and hypomyelination (Fancy et al. 2009). This effect is transient as CNS myelination ultimately catches up and appears normal in adult mice. Experimental demyelination performed on these transgenic mice results in a similar delay in oligodendrocyte differentiation and remyelination, without affecting OPC recruitment.
However, differences in the regulation of development and regeneration of myelin do occur; the transcription factor Olig1, although essential for developmental myelination (Xin et al. 2005), has a less redundant role in remyelination, where it plays a pivotal permissive role in OPC differentiation (Arnett et al. 2004). In contrast, notch signalling pathway, a negative (Wang et al. 1998) or positive (Hu et al. 2003) regulator of differentiation in development (depending on the ligand), is redundant during remyelination since conditional knockout of the notch1 gene in OPCs has no or a limited effect on remyelination (Stidworthy et al. 2004; Zhang et al. 2009).
Recently, the nuclear receptor retinoid X receptor-γ (RXRγ) has emerged a key positive regulator of oligodendrocyte differentiation directly from the analysis of remyelinating tissue (Huang et al. 2011). Microarray analysis of the separate stages of CNS remyelination in rats revealed that RXR is highly expressed in oligodendrocytes lineage cells during the differentiation phase of CNS remyelination. Transfection of cultured OPCs with siRNAs generated against RXRγ resulted in less morphologically differentiated oligodendrocytes. Analysis of RXRγ knockout mice that have received focal CNS demyelination resulted in the accumulation of undifferentiated OPCs and less mature oligodendrocytes in lesions. These results indicate that RXRγ regulates oligodendrocyte differentiation.
4.5.3 Inflammation and Remyelination
Several studies have provided compelling evidence for a key role of the inflammatory response to demyelination in creating an environment conducive to remyelination. The relationship between inflammation and regeneration is well recognised in many other tissues. However, its involvement in myelin regeneration has been obscured in a field dominated by the immune-mediated pathology of MS and its various animal models such as EAE, where it is unquestionably true that the adaptive immune response mediates tissue damage. Nevertheless, several descriptive studies using experimental models (Ludwin 1980) and MS tissue (Wolswijk 2002) have pointed to an association between inflammation and remyelination. The role of the innate immune response to demyelination in remyelination has become apparent in part through the use of non-immune-mediated, toxin-induced models of demyelination. Depletion or pharmacological inhibition of macrophages following toxin-induced demyelination leads to an impairment of remyelination (Kotter et al. 2005; Li et al. 2005), as does the absence of T cells (Bieber et al. 2003). The pro-inflammatory cytokines Il-1β and TNF-α, lymphotoxin-β receptor or MHCII have also been implicated as mediators of remyelination following cuprizone-induced demyelination (Mason et al. 2001; Plant et al. 2007; Arnett et al. 2001, 2003). A critical role played by phagocytic macrophages is the removal of myelin debris generated during demyelination since CNS myelin contains proteins inhibitory to OPC differentiation both in vitro and during remyelination (Baer et al. 2009; Kotter et al. 2006). The observation that macrophage activation enhances myelination by transplanted OPCs in the myelin-free retinal nerve fibre layer points to additional and as yet undefined regenerative factors produced by these macrophages (Setzu et al. 2006).
4.6 Demyelinated CNS Axons Can Also Be Remyelinated by Schwann Cells
CNS remyelination can also be mediated by Schwann cells, the myelin-forming cells of the peripheral nervous system; this occurs in several experimental animal models of demyelination as well as in human demyelinating disease (Snyder et al. 1975; Itoyama et al. 1983, 1985; Dusart et al. 1992; Felts et al. 2005). Schwann cell remyelination occurs preferentially where astrocytes are absent—for example, where they have been killed along with oligodendrocytes by the demyelinating agent (Blakemore 1975; Itoyama et al. 1985). Remyelinating Schwann cells within the CNS were generally thought to migrate into the CNS from PNS sources such as spinal and cranial roots, meningeal fibres or autonomic nerves following a breach in the glia limitans (Franklin and Blakemore 1993). In support of this idea, CNS Schwann cell remyelination typically occurs in proximity to spinal/cranial nerves or around blood vessels (Snyder et al. 1975; Duncan and Hoffman 1997; Sim et al. 2002a). However, recent genetic fate-mapping studies have revealed that very few CNS remyelinating Schwann cells are derived from PNS Schwann cells but instead the majority derive from OPCs (Zawadzka et al. 2010), revealing a remarkable capacity of these cells to differentiate into cells of neural crest lineage as well as all three neuroepithelial lineages (neurons, astrocytes and oligodendrocytes).
The implications of Schwann cell remyelination of CNS axons are unclear. While both Schwann cell and oligodendrocyte remyelination are associated with a return of saltatory conduction (Smith et al. 1979), their relative abilities to promote axon survival, a major function of myelin (Nave and Trapp 2008), have yet to be established. Thus, from a clinical perspective, we do not yet know whether OLP differentiation into Schwann cells has a beneficial or deleterious effect compared to oligodendrocyte remyelination.
4.7 Causes of Remyelination Failure
The efficiency of remyelination is affected by the non-disease-related factors of age and sex (Li et al. 2006; Sim et al. 2002b). These generic factors will have a bearing on the efficiency of remyelination regardless of the disease process involved and will be discussed first.
Like all other regenerative processes, the efficiency of remyelination decreases with age. This manifests as a decrease in the rate at which it occurs and is likely to have a profound bearing on the outcome of a disease process that in the case of MS can occur over many decades. The age-associated effects on remyelination are due to a decrease in the efficiency of both OPC recruitment and differentiation (Sim et al. 2002b). Of these two events, the impairment of differentiation is rate determining since increasing the provision of OPCs by the overexpression of the OPC mitogen and recruitment factor PDGF following demyelination in old mice does not accelerate remyelination (Woodruff et al. 2004). The impairment of OPC differentiation in ageing mirrors the failure of oligodendrocyte lineage differentiation associated with many chronically demyelinated MS plaques (Wolswijk 1998; Kuhlmann et al. 2008).
The basis of the ageing effect is likely to lie in age-associated changes in both the extrinsic environmental signals to which OPCs are exposed in remyelinating lesions and to intrinsic determinants of OPC behaviour. An impaired macrophage response in ageing, associated with a delay in expression of inflammatory cytokines and chemokines (Zhao et al. 2006), leads to poor clearance of myelin debris and the persistence of myelin-associated differentiation-inhibitory proteins. Changes also occur in the expression of remyelination-associated growth factors following toxin-induced demyelination that are commensurate with delays in OPC activation, recruitment and differentiation and are illustrative of age-associated environmental changes in the remyelination (Hinks and Franklin 2000). Both in vitro studies revealing age-associated changes in growth factor responsiveness of adult OPCs of different age (Tang et al. 2000) and in vivo studies demonstrating slower recruitment of transplanted old adult precursor cells compared to young adult-derived cells into precursor-depleted white matter (Chari et al. 2003) are indicative of intrinsic changes occurring with OPC during adult ageing. A recent study confirms these observations, revealing a critical age-associated change in the epigenetic regulation of OPC differentiation during remyelination (Shen et al. 2008). Differentiation of OPCs is associated with the recruitment of histone deacetylases (HDACs) to promoter regions of differentiation inhibitors (Marin-Husstege et al. 2002). In old animals, HDAC recruitment is impaired, resulting in prolonged expression of these inhibitors, delayed OPC differentiation and hence slower remyelination. This effect can be replicated following induction of demyelination in young animals with the use of the HDAC antagonist valproic acid. A key question relating to the development of remyelination therapies is the extent to which age-associated changes can be reversed. Intriguing experiments on skeletal muscle regeneration using the technique of heterochronic parabiosis provide clear proof of principle that poor regeneration in old animals can be rejuvenated (Conboy et al. 2005).
In addition to these generic factors, remyelination could also be incomplete or fail for disease-specific reasons. The strongest evidence for remyelination failure is provided by MS, and the subsequent discussion will specifically relate to this disease, although the issues discussed will be relevant to other diseases with a demyelinating component. Theoretically, remyelination could fail because of (1) a primary deficiency in precursor cells, (2) a failure of precursor cell recruitment or (3) a failure of precursor cell differentiation and maturation.
Early speculation on remyelination failure focussed on the first of these mechanisms, that is, the process of remyelination itself would deplete an area of CNS of its precursor cells so that subsequent episodes of demyelination occurring at or around the same site would fail to remyelinate due to a lack of OPCs. However, data from experimental studies indicate that OPCs are remarkably efficient at repopulating regions from which they have been depleted (Chari and Blakemore 2002) and that repeat episodes of focal demyelination in the same area (Penderis et al. 2003a) neither deplete OPCs nor prevent subsequent remyelination. The situation may be different, however, when the same area of tissue is exposed to a sustained demyelinating insult, where remyelination impairment seems to be due, at least in part, to a deficiency in OPC availability (Armstrong et al. 2006; Mason et al. 2004).
In the second mechanism, MS lesions fail to remyelinate not because of a shortage of available precursor cells but rather because of a failure of OPC recruitment: proliferation, migration and repopulation of areas of demyelination. Here, descriptions of demyelinated areas from which oligodendrocyte lineage (OL) cells are absent do indicate that this may account for failure of remyelination in at least a proportion of lesions. Why lesions should become deficient in OPCs is not clear, but one possibility is that they are direct targets of the disease process within the lesion. The identification of patients with antibodies recognising OPC-expressed antigens (NG2) supports this possibility (Niehaus et al. 2000). Failure of OPC recruitment into areas of demyelination may arise due to disturbances in the local expression of the OPC migration guidance cues semaphorins 3A and 3F (Williams et al. 2007a). In situations where OPCs need to be recruited into lesions from surrounding intact tissue, the size of the lesion will clearly have a bearing on the efficiency of remyelination, larger lesions requiring a greater OPC recruitment impetus than smaller ones, especially in ageing where older OPCs appear intrinsically less responsive to recruitment signals.
The best evidence at present supports the third mechanism, a failure of differentiation and maturation, as several sets of observations based on the detection of oligodendrocyte lineage cells within areas of demyelination indicate that this stage of remyelination is the most vulnerable to failure in MS. The presence of OPCs apparently unable to differentiate within MS lesions was initially shown with the OL marker O4 (Wolswijk 1998) and subsequently with NG2 (Chang et al. 2000), with PLP (to reveal pre-myelinating oligodendrocytes) (Chang et al. 2002) and with Olig2 and Nkx2.2 (Kuhlmann et al. 2008). Even though the density of OPCs within chronic lesions is on average lower than in normal white matter, the density can be as high as that in normal white matter or remyelinated lesions, showing that OPC availability is not a limiting factor for remyelination.
One possible explanation for this failure of differentiation is that chronically demyelinated lesions contain factors that inhibit precursor differentiation. First implicated was the notch-jagged pathway, a negative regulator of OPC differentiation: notch and its downstream activator Hes5 were detected in OPCs and jagged in astrocytes within chronic demyelinated MS lesions (John et al. 2002). However, the expression of notch by OPCs and jagged by other cells within lesions undergoing remyelination and, more informatively, the limited remyelination phenotype in experimental models following conditional deletion of notch in OL cells suggest that notch-jagged signalling is not a critical non-redundant negative regulator of remyelination (Zhang et al. 2009; Stidworthy et al. 2004). The ability of inhibitors of γ-secretase, an enzyme involved in the notch pathway, to enhance recovery following EAE might be indicative of an inhibitory role for notch signalling in remyelination, but is difficult to interpret given the additional expression of notch in the inflammatory effector cells.
Other potential inhibitory factors have been identified in other experimental and pathological studies. The accumulation of the glycosaminoglycan inhibitor of OPC differentiation hyaluronan within MS lesions may contribute to an environment within chronic lesions that is not conducive to remyelination by inhibiting OPC function via TLR2 signalling (Back et al. 2005; Sloane et al. 2010). The demyelinated axon itself has been implicated since demyelinated axons have been shown to express the adhesion molecule PSA-NCAM (Charles et al. 2002), which inhibits myelination in cell culture (Charles et al. 2000). The possibility that the properties of the OPC within areas of demyelination might be regulated by synaptic input from axons represents an exciting new development in the understanding of the complexity of regulatory factors that govern remyelination and by extension account for remyelination failure (Etxeberria et al. 2010).
While many studies in the last few years have concentrated on putative inhibitory signals to account for the failure of OPC to undergo complete differentiation within demyelinated MS plaques, an alternative explanation is that these lesions fail to remyelinate because of a deficiency of signals that induce differentiation. This hypothesis, based on the absence of factors, is difficult to prove but is consistent with a model of remyelination in which the acute inflammatory events play a key role in precursor activation and creating an environment conducive to remyelination (see above). While MS lesions are rarely devoid of any inflammatory activity, chronic lesions are relatively non-inflammatory compared to the acute lesions and constitute a less active environment in which OPC differentiation might become quiescent. Acute inflammatory lesions are characterised by reactive astrocytes that are the sources of many pro-remyelination-signalling factors (Williams et al. 2007b; Moore et al. 2011). In contrast, chronic quiescent lesions are characterised by scarring astrocytes that are transcriptionally quiet compared to reactive astrocytes. The scarring astrocyte is better viewed as a consequence of remyelination failure and not its cause. Thus, neither the reactive nor the scarring astrocytes (both confusingly contributing to astrogliosis) should be viewed as primary drivers of remyelination failure.
The two possibilities that remyelination failure reflects the presence of negative factors or the absence of positive factors are not, of course, mutually exclusive. Moreover, it has become apparent from many studies in recent years that there are a multitude of interacting factors, both environmental and intrinsic, that guide the behaviour of OL cells through the various stages of remyelination. Efficient remyelination may depend as much on the precise timing of action as on the presence or absence of these factors. In an earlier review, we articulated this in a model called the dysregulation hypothesis in which remyelination failure reflects an inappropriate sequence of events (Franklin and ffrench-Constant 2008; Franklin 2002; Fancy et al. 2011a). While the causes of remyelination failure in such a varied disease as MS are likely to be multiple, we still regard hypothesis as useful for understanding remyelination failure in the majority of cases.
Related posts:
 Immune Modulation and Repair Following Neural Stem Cell Transplantation
Immune Modulation and Repair Following Neural Stem Cell Transplantation
 Effects of Current Medical Therapies on Reparative and Neuroprotective Functions in Multiple Sclerosis
Effects of Current Medical Therapies on Reparative and Neuroprotective Functions in Multiple Sclerosis
 Development of Oligodendrocytes in the Vertebrate CNS
Development of Oligodendrocytes in the Vertebrate CNS
 A Peripheral Alternative to Central Nervous System Myelin Repair
A Peripheral Alternative to Central Nervous System Myelin Repair
 Exogenous Cell Myelin Repair and Neuroprotection in Multiple Sclerosis
Exogenous Cell Myelin Repair and Neuroprotection in Multiple Sclerosis
 Axonal Protection with Sodium Channel Blocking Agents in Models of Multiple Sclerosis
Axonal Protection with Sodium Channel Blocking Agents in Models of Multiple Sclerosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


