Fig. 5.1
Transplantation of oligosphere cells into md rats. Before transplantation, the oligosphere cells were transduced with retrovirus to express β-gal (a). Between 10 and 14 days after transplantation, a white streak of myelin was seen along the dorsal surface of the cord (b). The black dots are sterile charcoal marking the injection site. When the cord was stained with X-gal, the white streak turned to blue (c). Cross section of the cord indicated the location of the blue staining mainly in the dorsal funiculus but also in gray matter (d). Immunostaining of the transplanted cord showed PLP+ myelin in the dorsal funiculus with blue oligodendrocytes interspersing them (e, f). The inset in the lower right of (f) is the magnification of a “blue” oligodendrocyte with processes attaching to PLP+ myelin sheaths (arrow). Semithin sections demonstrated that a large number of myelin sheaths and blue cells were in the dorsal funiculus (g). Bar: 100 μm (from Zhang et al. 1998a)
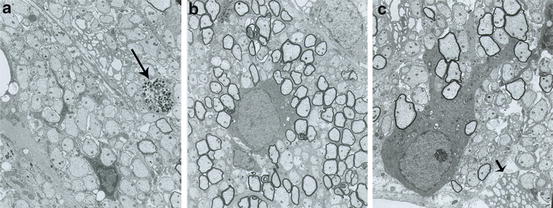
Fig. 5.2
Electron micrographs from representative myelinated areas in an md rat produced by rat neurospheres. (a) Adjacent to the transplant site, most of the axons are nonmyelinated. As in the uninjected mutant, no normal, native oligodendrocytes are present. On the far right in this field, a single degenerating axon is seen (arrow). (b) In a transplanted area, the majority of the axons are myelinated with a normal oligodendrocyte present. (c) Underneath the pia, a normal appearing oligodendrocyte has extensive processes leading to many myelinated fibers. Adjacent to this cell, the cytoplasm of an abnormal md oligodendrocyte is present (small arrow) with distended rough endoplasmic reticulum, a characteristic feature in md oligodendrocytes
While focal myelination by transplanted cells is easy to achieve, more global repair has also been documented in the myelin mutants. Studies of transplantation into the spinal cord of the sh pup showed more extensive myelination across the spinal cord at the grafting site and more rostral and caudal myelination to it (up to 20 mm) than in prior rodent studies (Archer et al. 1997) (Fig. 5.3). This was calculated to be ten times the volume of myelin achieved in the md rat. In the shi mouse, Mitome et al. (2001) reported extensive myelination in the brain and upper cervical cord by injecting NSCs into the lateral ventricles and cisterna magna. Kondo et al. (2005) demonstrated similar myelination in the brain of shi using OPCs but importantly showed that transplantation into the brain and spinal cord of the same mouse resulted in spatially separated areas of repair. This is critical to the potential application of transplantation to MS, as it proves that different strategic sites in brain and spinal cord could be remyelinated by more than a single transplant. Windrem et al. (2008) extended these results by transplanting OPCs into four sites, in the corpus callosum and cerebellar peduncles. For the first time, they showed that the brain could be almost completely myelinated by transplanted human glia and that this resulted in an improvement in the phenotype and extended survival (1 year or more) of 23 % of the grafted shi mice.
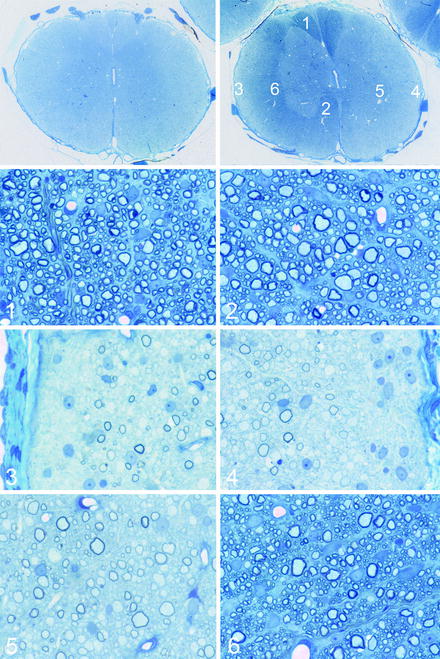
Fig. 5.3
Transplantation of neonatal mixed glial cell preparations into the shaking pup spinal cord. The montage illustrates the result of transplantations of such cells into a 7-day-old sh pup thoracic spinal cord, 8 weeks after transplantation. The top left figure shows a spinal cord segment 5 cm away from the transplant site and the top right figure, at the site of transplantation. At the site of transplantation, there is clearly evidence of myelin in the dorsal, lateral (left), and ventral column (right). Areas from the spinal cord (1–6) are shown in the lower subsequent figures. In 1, 2, and 6, there is complete myelination by the transplanted cells. In the lateral columns, the superficial left lateral column (3) and the entire right column (5) compared to the deep left lateral column (4) indicating extensive but incomplete migration and myelination by the transplanted cells
Many of the transplant studies in the myelin mutants have been performed in neonatal animals. As MS is a disease that affects the mature CNS, a question arises regarding the feasibility of the successful transplantation cells into the brain or spinal cord of adults, where the cues that drive myelination during development may be missing. Fortunately a number of experiments in different model systems have clearly shown that grafting cells into the mature CNS can result in myelination by the transplanted cells, including transplantation shi (Buchet et al. 2011; Mothe and Tator 2008) and the sh pup (Archer et al. 1997). In addition, this has been tested using both NSCs and OPCs. Indeed, transplanting cells into focally demyelinated lesions in the spinal cord or brain is usually performed in mature rodents, confirming observations in the older myelin mutant studies. Finally, confirmation of the ability to myelinate CNS axons throughout life comes from studies where NSCs or OPCs were grafted into the non-myelinated portion of the retina in both neonates (Ader et al. 2000; Setzu et al. 2004) and mature rats (Setzu et al. 2004).
Extending the spatial extent of remyelination by transplanted cells may be achieved by promoting the migration and division of transplanted cells. In all neural transplants, significant death of cells occurs acutely, and the division of surviving cells will be critical to success. It has been shown that both NSCs and OPCs may divide on engraftment (Milward et al. 2000; Windrem et al. 2008). It has also been shown that the division of OPCs can be increased by their co-transplantation with growth factor-producing cells. We have shown that an OPC cell line, CG-4, co-grafted in the md rat with neuroblastoma cells (B104), divided more than OPCs alone and hence occupied a greater area of neuropil (Milward et al. 2000) (Fig. 5.4). In addition, these studies demonstrated that the direction of migration can be influenced; when the two cells noted above were grafted at different sites, the OPCs migrated preferentially toward the B104 cells (Milward et al. 2000) (Fig. 5.4). More recently, we have shown that similar chemotactic attraction occurs when OPCs are transplanted adjacent to focal areas of inflammation (Wang et al. 2011b). The migration of transplanted cells through a non-myelinated neuropil, such as in the myelin mutants, may be relatively straightforward, but the ability of these cells to migrate through normal areas of white matter, for example, from one MS plaque to another, may present difficulties. Evidence suggesting that normal white matter does not support such migration has been reported (O’Leary and Blakemore 1997), yet a more recent study of transplanting human NSCs into demyelinated lesions of adult nude mouse spinal cord showed extensive migration, rostral and caudal to the transplant site and through areas of normal myelin (Buchet et al. 2011).
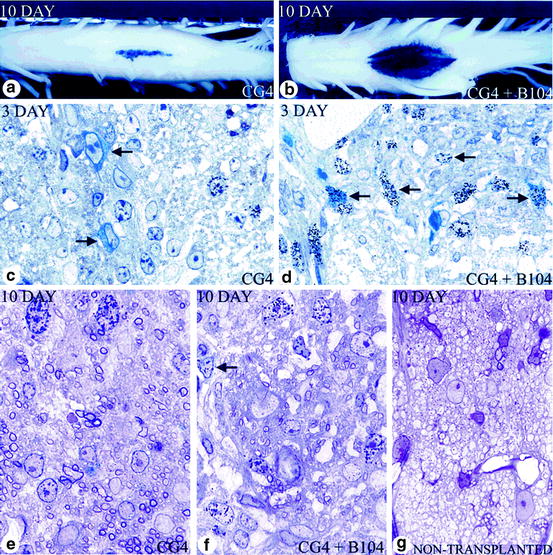
Fig. 5.4
Effects of cografting growth factor secreting cells on oligodendroglial progenitor cells in vivo. (a) When 50,000 CG-4 cells were injected alone, they were localized to the center of the dorsal column. (b) In contrast to (a), when an identical number of CG-4 cells were transplanted with 5,000 B104 cells, there was much greater lateral spread with more dense X-gal labeling of the spinal cord 10 days after transplantation. (c, d) At 3 days after transplantation in rats that received CG-4 cells alone (c) or CG-4 and B104 cells (d), more blue cells (CG-4) (arrows) are seen in animals that received the cograft. More of the blue cells in the cograft are labeled with silver grains, indicating greater division of CG-4 cells. At 10 days after transplantation, myelin formed by the transplanted cells was seen in both grafts (e, f) compared with the nontransplanted md rat, in which no myelin is present (g). However, more myelin was seen in those rats that received CG-4 cells alone (e) than in the cograft (f). In addition, more dividing cells were still seen in the cografted rats (f; seven in this field); many of those cells that were labeled with silver grains (e.g., arrow) showed thin blue perinuclear X-gal staining, (a–g) after X-gal histochemical stain; (c–g) 1-μm sections from rats labeled with [3H]-thymidine. Sections from X-gal-stained tissue and counterstained with toluidine blue (from Milward et al. 2000)
Migration of OPCs in vitro has been studied extensively, and the development and normal myelination of the CNS are dependent on their expression of key molecules and their interaction with the extracellular matrix (De Castro and Bribián 2005; Jarjour and Kennedy 2004). Remyelination may call for the extension of these interactions in vivo following transplantation of cells. In MS lesions, molecules such as semaphorin 3A and F (Piaton et al. 2011; Syed et al. 2011) and chemokines and their receptors (Kerstetter et al. 2009) may influence migration and in some cases the differentiation of OPCs in a positive or negative way. Transduction of NSCs with two molecules known to be involved in migration of OPCs in vitro, FGF2 and PSA-NCAM, has been studied to determine their effects on migration and differentiation both in vitro and in vivo. FGF2, a known mitogen of OPCs, was transduced into the OPC cell line, CG-4 (Magy et al. 2003), and shown to enhance proliferation and migration in vitro though not inhibiting differentiation. Transduction of NSCs with PSA-NCAM (Franceschini et al. 2004) or oligospheres (Decker et al. 2000; Vitry et al. 2001) and transplantation of these cells into shi brain extended the migration of the latter but not the former and, with its downregulation, the differentiation and myelination by other cells (Magy et al. 2003). It is not entirely clear however whether this cell manipulation significantly extended migration of the cells compared to controls. While these approaches are experimentally interesting and important, it seems likely that inducing expression of transgenes in human cells that are to be used in clinical trials would increase the difficulty in achieving Institutional Review Board and the Food and Drug Administration approval. A more detailed analysis of small molecules, cytokines, etc. that may promote endogenous remyelination is presented in Chap. 4 by Franklin et al.
As with the established data of cell transplantation into the myelin mutants, much is known and has been documented on the transplantation of cells into focal, chemically demyelinated lesions. This work has been pioneered by Blakemore and Franklin, and through many detailed, wide ranging studies, they have examined the glial cell–axonal interactions that occur in this model system (Blakemore et al. 1995a; Blakemore and Franklin 2000; Franklin and Blakemore 1997). An instant advantage over the myelin mutants is that these studies can be performed in adult animals which is not always possible in the myelin mutants, yet there are some disadvantages. For example, in animals where the spinal cord or brain is irradiated to prevent endogenous repair, it is only possible to study test animals up to 1 month after irradiation, as necrosis at the site will eventually occur. From the perspective of transplanting cells into MS lesions, a number of important issues have been highlighted by these studies and these include (1) as with endogenous remyelination, transplant-induced repair results in thin myelin sheaths. It remains to be shown however whether this is deleterious to long-term conduction in such axons and whether thin myelin remains neuroprotective (Franklin 1993); (2) OPCs are a better source of remyelination than adult oligodendrocytes (Crang et al. 1998), and their division is required for remyelination to occur; (3) astrocytes can be both helpful and inhibitory to exogenous cell remyelination (Blakemore et al. 2003; Franklin et al. 1991). Astrocytes are known to produce growth factors that play a key role in myelination in development (Moore et al. 2011). In demyelinated lesions where astrocytes are present, the success of remyelination by transplanted OPCs is dependent upon the “activation” status of astrocytes which will differ in acute and chronic demyelination (Blakemore et al. 2003). In regard to chronic MS lesions, the degree of gliosis is likely to be very important to the success of exogenous cells, if only through the inhibition of migration of cells in highly gliotic lesions; (4) OPCs transplanted into a normal neuropil do not migrate or survive well, but if the area is irradiated or in any way pathologic, then cells will survive (O’Leary and Blakemore 1997). This is an important issue as it will determine whether cells can migrate from one MS plaque to another through normal white matter. As they acknowledge, however, this issue is debated (Franklin and Blakemore 1997), and as discussed earlier here, other studies including a recently published report (Buchet et al. 2011) would appear to suggest that in certain experimental situations, this can occur. It was also noted that transplanted glial cells migrated over greater distances and remyelinated axons faster than endogenous cells (Blakemore et al. 2000). This implies that inhibitory signals for endogenous OPCs in MS plaques may not interfere with transplanted exogenous cells; hence one of the proposed major hurdles in MS may be unfounded; (5) chronically demyelinated tissue may not be permissive for exogenous cell repair. Thus, chronic plaques may need to be “primed” by promotion of inflammation at the site, for transplanted OPCs to differentiate and myelinate (Foote and Blakemore 2005); (6) remyelination protects demyelinated axons from degeneration. In studies in cuprizone-treated mice, irradiation of the brain to create persistent demyelination resulted in significant axon death which was prevented by transplantation of labeled NSCs into the lateral ventricles (Irvine and Blakemore 2008). Protection of axons was attributed to their remyelination by the transplanted cells. This is an important observation as it confirms the neuroprotective properties of remyelination therapy; (7) tissue matching of allografts may not be essential for graft survival. Adult oligodendrocytes can be induced to express major histocompatibility (MHC) class I or II except if induced with IFN-γ in vitro. However, OPCs do not express either in vitro or in vivo after transplantation (Tepavcevic and Blakemore 2006). Mixed glial cell transplants (including astrocytes and microglia) are rejected after 1 month but pure OPCs are not (Tepavcevic and Blakemore 2005). This highlights the importance of purifying cell preparations prior to grafting and using primarily OPCs in allograft transplants. However, it should be noted that subsequent rejection of allografted cells may not be entirely negative as the resultant inflammation may provoke endogenous remyelination (Blakemore et al. 1995b). Thus in chronic MS plaques where OPCs are present but non-differentiating, the rejection of transplanted cells may trigger endogenous remyelination by the persisting OPCs and underscore the view that exogenous and endogenous repair are not mutually exclusive.
5.4 What Cells Can Be Used as an Exogenous Source of Remyelination?
Without doubt, one of the most important issues on the transplantation of cells into MS lesions is the choice and source of cells to be used (Duncan 2008; Duncan et al. 2008). This section will deal with cells of the oligodendrocyte lineage that may be candidates for therapeutic consideration, while this will be followed by a chapter on the role that Schwann cells may have in remyelination in MS. The lineage of the oligodendrocyte is one of the most studied of all the cells of the CNS (see Chap. 1 by Miller). In dissecting apart the lineage from the perspective of identifying stages that could be used for exogenous repair, it is worthwhile considering the ideal characteristics of cells for use in remyelinating MS lesions. These cells should be (1) able to migrate long distances, (2) capable of many divisions but do not become transformed, (3) able to myelinate many axons, (4) unlikely to provoke an immune response or this can be prevented, and (5) available in large numbers and can be cryopreserved.
Based on these characteristics, what is known about the development and differentiation of oligodendrocytes and their precursors that will fulfill the criteria required? While it is certain that the cells to be used in the first clinical trials in MS will be of human origin, much of what is known about oligodendrocytes comes from studies on the rodent CNS (see Chap. 1 by Miller). As can be seen in Fig. 5.5, the oligodendrocyte lineage is complex with multiple, defined, and likely, yet to be defined stages, originating as all cells from the embryonic stem cell (ESC) and developing through the NSC to the mature myelinating oligodendrocyte. Much of this information has been derived from in vitro studies and correlated and confirmed where possible in vivo using both wild type and transgenic mice and rats. In summary, the development of oligodendrocytes from the first cells of the embryo, the ESCs, requires the critical temporal expression of a wide range of transcription factors (Nicolay et al. 2007), surface markers, and myelin genes (Fig. 5.5) (Buchet and Baron-Van Evercooren 2009). While many of the transcription factors may not be essential for development, Olig1/2, Sox 10, and Nkx2.2 are the most important and require signals such as sonic hedgehog (SH) for their correct spatial–temporal expression. The membrane proteins, NG-2, and the platelet-derived growth factor receptor-α (PDGF-Rα) are markers of the OPC as well as the sulfatide O4. Premyelinating then myelinating oligodendrocytes will express O4 then O1 (Galc), also surface markers, followed by the expression of myelin genes in mature cells, such as MBP, PLP, myelin-associated glycoprotein (MAG), and MOG. As the cell differentiates, it progresses from the classic bipolar OPC (at neonatal stages) to the early multipolar, immature oligodendrocyte to the mature oligodendrocyte with multiple processes and membrane sheets (Fig. 5.6). This in vitro scheme is also likely similar or identical to oligodendrocyte development in vivo. The identification of cells that will myelinate or remyelinate axons on transplantation into the CNS is most assured if they are shown to follow such a pattern in tissue culture prior to grafting.
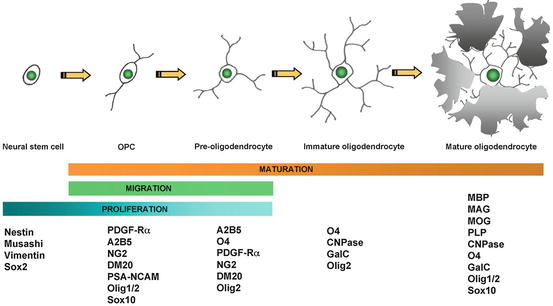
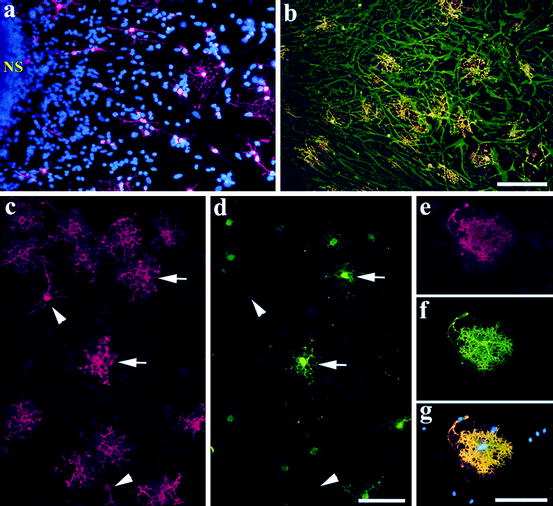
Fig. 5.5
Schematic diagram of the four-step maturation of neural stem cells toward the oligodendroglial lineage. Oligodendrocyte progenitor cells (OPCs), preoligodendrocytes, immature oligodendrocytes, and mature (myelinating) oligodendrocytes can be distinguished by their increasingly complex morphology; by their proliferating, migrating, and myelinating potentials; as well as by their expression of a wide range of well-defined markers. These markers may not be identical between rodents and humans (modified from Buchet and Baron-Van Evercooren 2009 with permission)
Fig. 5.6
Differentiation of human oligodendroglia from neural precursor cells. (a) A neurosphere (NS; passage 3) was cultured in the differentiation condition for 7 days in vitro (DIV) and stained with O4, showing that O4 oligodendroglia emerged from the neurosphere. Note the morphological transition from bipolar cells adjacent to the sphere to more branched oligodendroglia in the periphery. (b) A similar culture (from passage 7) for 15 DIV was stained with O1 (in red) and GFAP (in green). A 23-day-old differentiating culture was stained with O4 (c) and MBP (d) showing that the well-branched (arrows) but not the morphologically simple (arrowheads) oligodendroglia expressed MBP. (e–g) An oligodendrocyte that grew outside of the astrocytic layer and exhibited membrane-like structure. Cell nuclei in (a) and (g) were stained with DAPI (in blue). Bar = 100 μm (from Zhang et al. 1999)
5.4.1 Embryonic Stem Cells
As the pluripotent cell that gives rise to all cells of the body and is capable of providing an almost infinite number of cells, ESCs have been explored as a source of oligodendrocytes. In the original study of Brüstle et al. (1999), three mouse ESC lines were grown in the presence of leukemia inhibiting factor (LIF) which was then withdrawn with resultant formation of embryoid bodies (EBs). Using conditions that favor differentiation toward NSCs with media containing growth factors, then with addition of FGF2, epidermal growth factor (EGF), and PDGF, a population of cells that were positive for the surface marker, A2B5, was isolated. Upon removal of the growth factors, around 35 % of the cells differentiated into either oligodendrocytes or astrocytes (Brüstle et al. 1999). Transplantation of the cells prior to growth factor withdrawal demonstrated their oligodendrocyte differentiation and myelination in the md rat as well as their differentiation in vivo into astrocytes (Brüstle et al. 1999). A second, similar study published 1 year later showed evidence of oligodendrocyte differentiation of cells derived from mouse ESCs, in areas of chemically induced demyelination in the spinal cord and also following transplantation into the shi spinal cord (Liu et al. 2000). An increase in the differentiation of NSCs derived from mouse ESCs to oligodendrocytes has been reported when they were transduced to express an interleukin-6/soluble interleukin-6-receptor fusion protein (Zhang et al. 2006). This also increased myelination when these cells were transplanted into shi mice. Induction of Olig2 in mouse ESCs promoted the differentiation to oligodendrocytes during gliogenesis in the absence of sonic hedgehog, previously shown essential for oligodendrocyte differentiation from NSCs (Du et al. 2006).
Despite being isolated many years after mouse ESCs, human ESCs (hESCs) have taken center stage since 1998 (Thomson et al. 1998) as the potential source of cells for much of regenerative medicine and in particular here, as a source of NSCs that could be differentiated into neurons or glia and used in the therapy of neurodegenerative disease (Lindvall and Kokaia 2006). In 2001, Zhang et al. provided proof that NSCs subsequently OPCs then oligodendrocytes could be derived from hESCs (Zhang et al. 2001). Like mouse ESCs, the hESCs were cultured with the development of embryoid bodies, though in the presence of FGF2. Within these structures, neural rosettes develop that can be manipulated to high purity, NSC-containing cultures, with the production finally of neurons, astrocytes, and a minor proportion of cells of the oligodendrocyte lineage (5 % or less (Zhang et al. 2001)). In essence, this culture system is long (up to 3 months) and complex involving the induction of NSCs, patterning of Olig2 progenitors, differentiation of Olig2/Nkx2.2 expressing pre-OPCs, then the generation of OPCs. This calls for sequential culturing in chemically defined media, followed by exposure to retinoic acid and SH then FGF2. The differentiation of OPCs is then dependent on more standard protocols for these cells (Hu et al. 2009). This culture protocol can lead to up to 80 % of cells becoming OPCs, with 40 % becoming immature oligodendrocytes yet few differentiating to pure MBP-expressing mature oligodendrocytes (Gumpel et al. 1983). This culture system underscores the difficulty of isolating significant numbers of mature, human oligodendrocytes yet large numbers of OPCs. Other protocols have been reported that documents NSC (Joannides et al. 2007; Reubinoff et al. 2001) derivation from hESCs.
In contrast to the reports described above, the method of isolating mature oligodendrocytes from hESCs of Keirstead and colleagues (Nistor et al. 2005) resulted in a purity of up to 95 % of cells expressing the mature oligodendrocyte marker Galc. Perhaps the major difference between this and other protocols is the selection of cell aggregates (spheres) growing in the presence of retinoic acid selected for their yellow color (perhaps indicating viability), followed by subsequent growth in retinoic acid-free media, leading to the purified cultures noted above. More recent studies on oligodendrocyte differentiation from NSCs derived from hESCs have adopted certain differences in the culture protocol to maximize the generation of OPCs (Izrael et al. 2007; Kang et al. 2007). Both produced enriched populations of OPCs and in the first study, the addition of noggin, the bone morphogenetic protein antagonist, increased the number of mature oligodendrocytes (Izrael et al. 2007).
The clinical future for pluripotent stem cells may rest however with induced pluripotent cells (iPSCs) as in principle, they can be generated on an individual patient basis and cells derived from them are used as autologous grafts; hence immunosuppression would not be necessary (Kiskinis and Eggan 2010; Koch et al. 2009). Recent results from generating mouse iPSCs from skin fibroblasts are promising as the differentiation to NSCs and then OPCs resulted in around 18 % of cells becoming mature oligodendrocytes expressing MBP (Czepiel et al. 2011). Transplantation of OPCs derived in this fashion into chronic cuprizone lesions in the corpus callosum resulted in scattered MBP-positive areas (Czepiel et al. 2011). As with hESCs, the differentiation of human iPSCs toward OPCs and oligodendrocytes is not straightforward, and using different reprogramming techniques resulted in variable differentiation efficiencies (Hu et al. 2010). The current intense search and refinement in reprogramming methods of adult somatic cells (Amabile and Meissner 2009; Yamanaka and Blau 2010) and improvement of differentiation protocols suggest that iPSCs may become the eventual source of human OPCs and oligodendrocytes for transplantation, though perhaps not always on an individual patient basis (Kiskinis and Eggan 2010). A caveat however is that the epigenetic events of reprogramming can cause DNA damage, and the significance of these events may have to the translational use of the cells remains to be decided. Alternately, the direct conversion from adult somatic cells to OPCs, skipping the conversion to pluripotent cells as has been shown for the conversion of mouse and human fibroblasts to neurons (Pang et al. 2011), will also be a target.
5.4.2 Neural Stem Cells
Since the report of Reynolds and Weiss in 1992 of the isolation and culture of multipotent NSCs in the presence of EGF and FGF2, as free-floating spheres called neurospheres, from the adult striatum of mice (Reynolds and Weiss 1992) and subsequently embryonic brain (Reynolds and Weiss 1996), this protocol has become the optimal method of studying NSCs and their isolation for therapeutic purposes (Pastrana et al. 2011). Removal of the growth factors leads to the cells differentiating into neurons, astrocytes, and oligodendrocytes, though a minority of cells become oligodendrocytes in vitro (Reynolds and Weiss 1992). This pattern of differentiation however can be influenced by the in vivo environment into which they are grafted if cues driving differentiation toward a gliogenic fate and particularly toward an oligodendrocyte need are present. In a study on the transplantation of rat NSCs into the md rat, Hammang et al. (1997) showed that these cells preferentially differentiated into OPCs then oligodendrocytes which myelinated significantly large areas of the spinal cord at the site of injection. Since then, numerous other studies have confirmed that NSCs derived from the fetal and adult CNS can give rise to myelinating oligodendrocytes (Mothe et al. 2008; Mothe and Tator 2008). NSCs have been used quite extensively in spinal cord injury models where functional recovery has been associated with their development into both oligodendrocytes and Schwann cells. In addition, mouse NSCs (Karimi-Abdolrezaee et al. 2006) or those derived from ESCs (Kumagai et al. 2009) or iPSC (Tsuji et al. 2010) have also been used in spinal cord injury models where they took part in remyelination. Human NSCs have also been shown to give rise to oligodendrocytes in vitro and in vivo following lysolecithin demyelination of the corpus callosum (Neri et al. 2010). In addition, mouse iPSC-derived NSCs have also been shown to remyelinate axons in a spinal cord injury model (Tsuji et al. 2010). However, NSCs may have pleiotropic effects on transplantation into the CNS and not only be capable of remyelination. Thus when transplanted into EAE models where they ameliorate disease, this is thought to be principally through immunomodulation (Pluchino et al. 2003, 2005), or promotion of endogenous remyelination as shown in chronic cuprizone intoxication (Einstein et al. 2009). If their primary use is to repair myelin however, it remains undecided whether NSCs are a poorer source of OPCs than using OPCs directly (Smith and Blakemore 2000). We would argue that the study of Hammang et al. (1997) and subsequent reports (Mothe et al. 2008; Mothe and Tator 2008) have shown that NSCs are a viable source of cells for remyelination, though a head-to-head comparison of NSCs vs. OPCs in an experimental model would be required to determine if one or other leads to greater myelination. A concern that NSCs, which by definition are multipotent, will give rise to ectopic neurons or astrocytes in white matter has not been found to be excessive or inhibit myelin repair.
5.4.3 Oligodendrocyte Progenitor Cells and Oligodendrocytes
Conventional wisdom would suggest that OPCs are the best cell for myelin repair as they fit many of the criteria defined earlier. They are migratory, mitotic, and known to differentiate into oligodendrocytes. While this is self-evident, they may also be the source of remyelinating Schwann cells (Zawadzka et al. 2010) in astrocyte-free areas. Many studies however have confirmed their differentiation into myelinating oligodendrocytes in vivo following transplantation.
The OPC or as it was originally known, the O-2A cell, is one of the most studied cells of the CNS. Its acquisition from the CNS, and as used by Raff, Noble, Barres, Miller, and Richardson and others who studied it in vitro, was from the dissociation and culture of mixed glial cell preparations from the optic nerve or brain. Pure populations of OPCs, neonatal, and adult OPCs have been isolated by FACS, immunopanning, or magnetic bead sorting using antibodies to appropriate stage markers such as A2B5 (e.g., Shi et al. 1998), PSA-NCAM (e.g., Windrem et al. 2008), O1 (e.g., Duncan et al. 1992), and more recently CD140a (Sim et al. 2011). The limitation of these methods of isolating OPCs for transplantation has been in the numbers of cells generated from some of the protocols in terms of what would be required for human therapeutic application and in the complexity of the isolation procedure. An alternative strategy has been to generate OPC cell lines, though the downside of using, for example, SV40 transduction and immortalization is in the potential for transformation and tumor growth (Barnett et al. 1993; Trotter et al. 1993). However this approach resulted in the demonstration of extensive myelination by a retrovirally immortalized O-2A cell line (Groves et al. 1993). Perhaps the most useful OPC cell line has been CG-4, which was produced by growth factor expansion of OPCs (Louis et al. 1992). In culture, CG-4 cells differentiate into almost pure oligodendrocytes with few astrocytes though increasing serum in the media results in pure astrocyte populations. When transplanted into either the md rat or focal, glial-deficient demyelinated areas, CG-4 cells will give rise to myelinating oligodendrocytes (Franklin et al. 1996; Tontsch et al. 1994) and some astrocytes (Duncan 1996; Franklin et al. 1995).
In order to generate a greater supply of OPCs, Avellana-Adalid et al. (1996) developed a technique to isolate OPCs from neonatal rat brain by producing clusters of free-floating cells called oligospheres, purified by removal of astrocytes and oligodendrocytes by selective adhesion. Exposure to conditioned media from the B-104 neuroblastoma cell line resulted in a pure populations of OPCs which in low serum became pure oligodendrocytes while in high serum, astrocytes (Zhang et al. 1998a, b). These OPCs made myelin on transplantation into the shi mouse. A similar strategy was performed with mouse brain (Vitry et al. 1999). We chose a different method of purifying expandable collections of OPCs, isolating cells from the neonatal striatum of rats and canines, and culturing them initially as neurospheres in the presence of EGF and FGF2. By the gradual substitution of these growth factors with B-104 conditioned media, the cells switched to the oligodendrocyte lineage with over 95 % of cells differentiating into immature oligodendrocytes (Zhang et al. 1998a, b, 1999, 2000).
As noted at the beginning of this section, the cells that will be used in the first trials will be human. Thus, does what we know of rodent and higher animal oligodendrocyte development apply to humans? In short, similar principles apply to the differentiation of NSCs and OPCs in the human lineage as illustrated in Fig. 5.6. Indeed the maturation of human oligodendrocytes from neurospheres illustrates the classic features of OPC development toward the mature oligodendrocyte (Fig. 5.6). However there are significant differences between animal and human cells that lead to continued challenges in isolating sufficient OPCs for clinical use. A major gap in knowledge is a lack of known mitogens of the human OPC. Likewise, seemingly arcane yet potentially important differences, such as human O4-positive cells being non-mitotic, unlike rodent cells (Zhang et al. 2000) may be important. At present therefore, the generation of large numbers of human OPCs (or NSCs) for human clinical trial would seem to be most likely achievable using pluripotent cells as the source.
5.5 How Can Cells Be Tracked After Transplantation and Myelination Monitored?
It will be important to be able to track cells after grafting to determine whether they remain within the targeted lesion or migrate away to other sites. This question has been explored experimentally by labeling either NSCs, OPCs, Schwann cells, or OECs with superparamagnetic iron particles and following them by MRI (Bulte et al. 1999, 2001). Successful monitoring of cells has been achieved on transplantation into the md and Long Evans shaker (les) rats (Bulte et al. 1999, 2001), in focal demyelinated lesions (Dunning et al. 2004) and in EAE (Ben-Hur et al. 2007; Muja et al. 2011; Politi et al. 2007). It has been shown that labeling of cells does not interfere with their myelinating function (Bulte et al. 1999). It is also possible to follow cells in the mouse brain that express the luciferase gene using bioluminescence (Sher et al. 2009), but this is less likely to be used in patients because of the genetic manipulation of the cells and the technical limitations in detecting labeled cells in larger brains.
Of equal importance to following the transplanted cells will be monitoring remyelination in vivo. The primary method to achieve this will be with MRI, but the definitive MRI parameters of remyelination are still debated and require definitive data from an experimental system. Nonetheless, pre- and posttransplant MRIs, using magnetic transfer ratios, diffusion tensor imaging, and the newer method imaging “myelin water fraction” (Laule et al. 2007) will still provide the baseline information. Beyond MRI imaging, PET scanning using myelin-specific radiotracers (Stankoff et al. 2006; Wang et al. 2011a) has been used to follow myelination in development and in myelin mutants. A further advance using near-infrared imaging of myelination using a fluorescent probe that binds to myelin has been reported and studied in cuprizone-induced demyelination and remyelination (Wang et al. 2011a). However with both this and PET imaging, it remains to be proven that the techniques have the resolution to accurately image remyelination.
5.6 Do Exogenous Cells Remyelinate Axons in Inflammatory Disease?
There is growing interest in the role of inflammation in remyelination (Arnett et al. 2003; Hohlfeld 2007; Kondo et al. 2011; Popovich and Longbrake 2008; Setzu et al. 2006). Certainly endogenous remyelination occurs at the time of inflammatory cell infiltrate into the CNS, and a positive role of certain cytokines, once thought harmful to repair, has been postulated (Arnett et al. 2003). Likewise in experimental disorders, it has been shown that inflammation can promote both myelination (Setzu et al. 2006) and remyelination (Foote and Blakemore 2005). Myelination by oligodendrocytes derived from mouse ESCs has also been demonstrated in a focal, inflammatory model in the adult rat spinal cord (Perez-Bouza et al. 2005). The function of transplanted OPCs or other cell types has been most studied in EAE with some surprising results, most of them positive. While the original goal may have been to use transplanted cells to remyelinate axons, it has been shown that they may have pleiotropic effects. An important finding on transplanting cells into the CNS of animals with EAE is that both animal and human cells survive and that inflammation promotes their migration (Einstein et al. 2003; Tourbah et al. 1997) (Fig. 5.7). Confirmation of this has been reported following transplantation of OPCs adjacent to focal areas of inflammation created by injection of zymosan in the spinal cord (Wang et al. 2011b). Indeed, OPCs grafted adjacent to the lesions preferentially migrated into the lesion where they survived suggesting chemotactic signals in the lesion (Wang et al. 2011b) and some differentiated into myelinating oligodendrocytes.
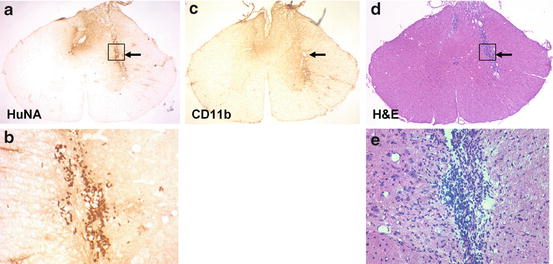
Fig. 5.7
Human cells survive after transplantation into areas of inflammation. Four weeks after transplantation of a glial-restricted human precursor cells (264) into Dark Agouti rats with EAE, human cells are indentified by immunolabeling for human nuclear antigen (HuNA) (a, b). They are located in areas of inflammation as noted in adjacent H + E stained sections (d, e) and in areas of microglial activation (OX-42) (c) (prepared by Dr. E. Larsen)
In a series of detailed experiments from Ben-Hur and colleagues, transplantation of NSCs into the lateral ventricles of mice with EAE demonstrated that encephalitis promoted the migration of the cells into the brain parenchyma (Einstein et al. 2003, 2006a–c). The cells differentiated into glial cells where they decreased inflammation and lessened axon loss (Aharonowiz et al. 2008; Einstein et al. 2006b). More recently, the same group showed that when NSCs were transplanted into the ventricles of mice treated with cuprizone, where there is a marked microglial response but no T-cell infiltration, the cells promoted endogenous OPCs to remyelinate axons in the corpus callosum. The transplantation of NSCs by a combined intravenous and intrathecal approach has also been shown to ameliorate EAE in a mouse model (Pluchino et al. 2003, 2005). These important experiments concluded that improvement was primarily through peripheral immunomodulation by the NSCs and not through remyelination. These data are discussed in greater detail by Martino and Ben-Hur (Chap. 7).
Transplantation of NSCs or glial-committed progenitors in mice with inflammatory demyelination resulting from MHV also resulted in lessening of disease, remyelination, and axonal protection. Firstly it was shown that transplanted cells survived and migrated within the demyelinated, inflamed spinal cord (Totoiu et al. 2004). These mice showed behavioral improvement associated with extensive remyelination and sparing of axons (Totoiu et al. 2004). In a follow-up study, they demonstrated that the remyelination was not associated with a decrease in inflammation as measured by T-cell and macrophage infiltration and expression of proinflammatory cytokines such as TNF-α (Hardison et al. 2006). In neither of these studies were they able to determine whether the remyelination resulted from the transplanted cells or from endogenous repair. The same group transplanted OPCs derived from hESCs into MHV-induced demyelinated spinal cord, but despite treatment with high-dose cyclosporine A, the cells were rejected after 2 weeks (Hatch et al. 2009). However extensive remyelination was seen at the site, and it was suggested to have resulted from an endogenous OPC response, stimulated by the xenograft rejection (Hatch et al. 2009). Finally, myelination by transplanted OPCs into the non-myelinated retina was enhanced by concurrent injection of zymosan which produced focal inflammation (Setzu et al. 2006), thus adding to the observations summarized above that highlight the importance of inflammation in repair.
5.7 How Is Functional Recovery Evaluated After Transplantation?
Evaluation of functional recovery after the transplantation of myelinating cells in animal models is documented by multiple levels of evidence (Zhang and Duncan 2000). The end goal of course is for MS patients receiving such a therapy to show clinical improvement. Evidence of functional improvement from the animal models will therefore provide assurance of potential success in human trials and is based on (1) the restoration of the myelin sheath (perhaps still thin) with normal nodes of Ranvier, (2) evidence that these structures have restored impulse conduction, and (3) evidence of behavioral improvement, for example, in muscle strength/movement and vision.
5.7.1 Myelin Sheath Thickness and Nodes of Ranvier
Remyelination both by endogenous and exogenous cells usually results in the formation of thinner-than-normal myelin sheaths around demyelinated/dysmyelinated axons. However, these myelin sheaths are capable of almost normal impulse conduction (Smith et al. 1981; Utzschneider et al. 1994). The reason that myelin sheaths remain thin on remyelination is not known though it may require axons to be growing for restoration of normal sheath thickness to occur. Certainly, transplantation into neonates may result in normal myelin sheaths after a prolonged period (Windrem et al. 2008). Nerve conduction is dependent on the formation of normal nodes of Ranvier. The node of Ranvier is defined by ultrastructural specializations at the node and paranode and a normal molecular architecture at these sites (Poliak and Peles 2003). A transplant study in the md rat showed normal axoglial junctions at the node, both by routine ultrastructural evaluation and freeze fracture (Rosenbluth et al. 1993) in areas of transplant myelin. Two reports of transplantation of rodent NSCs (Eftekharpour et al. 2007) or human OPCs (Windrem et al. 2008) into the shi examined the distribution of voltage-gated sodium and potassium channels, as well as another important paranodal molecule, contactin-associated protein (Caspr), in repaired areas, at nodes and paranodal areas. These studies confirmed that transplanted glia and the myelin made by these cells resulted in normal formation of nodes of Ranvier, thus providing the structural basis of normal impulse transmission.
5.7.2 Restoration of Impulse Conduction
The first proof that myelin produced by transplanted cells came from ex vivo studies on the spinal cords of md rats (Utzschneider et al. 1994). These studies clearly showed that the nerve conduction velocity as well as other physiological parameters were restored through the transplant site, in contrast to non-myelinated areas rostral and caudal to it (Utzschneider et al. 1994). The transplant site was grossly visible in the dorsal column of the isolated spinal cord allowing placement of stimulatory and recording electrodes at desired sites (Fig. 2 in Duncan and Milward 1995). In two studies of conduction in grafted areas of the CNS in the shi mouse following transplantation, conduction velocity was also determined to be increased, though not normal in the brain (Windrem et al. 2008) and spinal cord (Eftekharpour et al. 2007). Two studies on the physiological effect of transplanted cells in inflammatory disease have been reported. In the first, human NSCs were injected into either the cisterna magna or the tail vein of marmosets that had been immunized with MOG peptide resulting in development of EAE (Pluchino et al. 2009). Although there were improvements in central conduction following transcranial stimulation in transplanted marmosets, it is not certain that this resulted from remyelination as was shown histologically, and the authors suggested that clinical improvement related to the immunomodulatory effects of the NSCs. In the second study, transplantation of glial-restricted precursors (Walczak et al. 2011) was carried out into focal inflammatory lesions in the spinal cord. Evaluation of somatosensory-evoked potentials in the transplanted group showed a decrease in latency. Whether this was related to remyelination by the transplanted cells is also not entirely clear.
Related posts:
 Immune Modulation and Repair Following Neural Stem Cell Transplantation
Immune Modulation and Repair Following Neural Stem Cell Transplantation
 Effects of Current Medical Therapies on Reparative and Neuroprotective Functions in Multiple Sclerosis
Effects of Current Medical Therapies on Reparative and Neuroprotective Functions in Multiple Sclerosis
 Development of Oligodendrocytes in the Vertebrate CNS
Development of Oligodendrocytes in the Vertebrate CNS
 A Peripheral Alternative to Central Nervous System Myelin Repair
A Peripheral Alternative to Central Nervous System Myelin Repair
 Imaging of Demyelination and Remyelination in Multiple Sclerosis
Imaging of Demyelination and Remyelination in Multiple Sclerosis
 Axonal Protection with Sodium Channel Blocking Agents in Models of Multiple Sclerosis
Axonal Protection with Sodium Channel Blocking Agents in Models of Multiple Sclerosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


