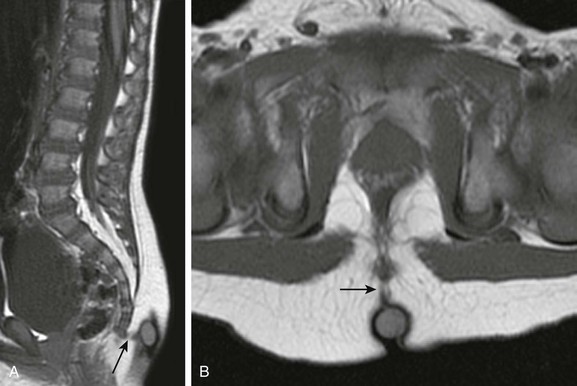
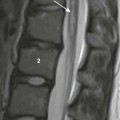
Chapter 43 Concurrent with the neural tube folding during primary neurulation, spinal cord development below the caudal neuropore commences within the pluripotent tissue at the caudal eminence in the process of secondary neurulation. The initially solid cell mass canalizes and becomes contiguous with the rostral neural tube that was formed by primary neurulation. By day 48, a transient ventriculus terminalis appears in the future conus. If this persists after birth, it is noted incidentally as a normal variant ventriculus terminalis (“fifth ventricle”), usually of no clinical significance (see Chapter 40). Failure of proper secondary neurulation leads to caudal spine anomalies in the caudal regression, tethered cord, or sacrococcygeal teratoma (SCT) spectra in addition to terminal myelocystocele and anterior sacral meningocele (ASM). Premature dysjunction of the neural tube from overlying ectoderm permits perineural mesenchyme to access the neural groove and ependymal lining. This mesenchyme differentiates into fat and prevents complete neural tube closure, resulting in skin-covered lipomatous malformations with or without posterior spinal dysraphism. The most commonly observed anomalies are lipomyelocele (LMC), LMMC (Fig. 43-1), and intradural spinal lipomas (Fig. 43-2). Figure 43-1 Lipomyelomeningocele. Figure 43-2 Juxtamedullary (subpial) spinal lipoma. In contrast to lipomatous malformations, anomalies that result from nondysjunction occur when the neural tube fails to dissociate from adjacent cutaneous tissue. The simplest and least extensive variation is the dorsal dermal sinus, which occurs when a single connection persists and forms a fibrous cord from a skin dimple to the dural sac, conus, or central spinal cord canal. It is important to distinguish dermal sinus from its clinically asymptomatic mimic, simple coccygeal dimple (Fig. 43-3). In this mimic, the low sacral or coccygeal sinus originates from a low skin dimple and attaches to the coccyx via a short fibrous tract. These dimples are nearly always found within the intergluteal cleft, never communicate with the spinal canal, and require no treatment. Simple coccygeal dimples are the most common reason for newborn spinal ultrasound imaging.
Congenital Abnormalities of the Spine
Embryology and Developmental Anatomy
Abnormalities of Primary Neurulation
Premature Dysjunction
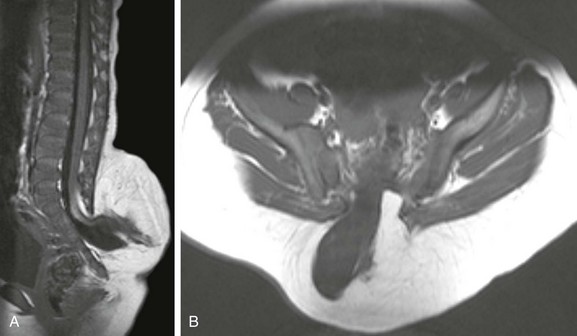
Sagittal (A) and axial (B) T1-weighted magnetic resonance (MR) images demonstrate a typical lipomyelomeningocele, with a low-lying cord tethered into a large lipomatous malformation contiguous with the subcutaneous fat through a posterior dysraphic defect.
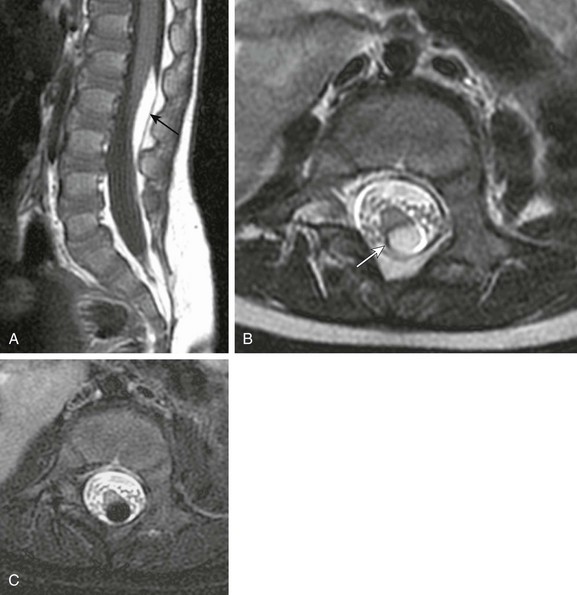
A, Sagittal T1-weighted magnetic resonance (MR) image shows a small subpial intradural lipoma (arrow) adherent to the dorsal conus surface. B, Axial T2-weighted MR image confirms direct contiguity of the neural placode with the lipoma. Note chemical shift artifact (arrow) in the frequency encoding direction, indicating fat. C, Axial fat-saturated T2-weighted MR image confirms fat content by homogeneous signal loss within the lipoma.
Nondysjunction
Congenital Abnormalities of the Spine