Chapter 10 Clinical Presentation and Etiologies: The external and middle ear both are derived from the branchial apparatus, in contrast to the inner ear, which is of neuroectodermal origin. As a result, dysplasia of the external auditory canal (EAC) is commonly associated with a malformed auricle and anomalies of the middle ear and the mandible. These children usually present with a malformed pinna and conductive hearing loss. Inner ear anomalies occur in 20% of children with aural atresia and are more likely to involve the vestibular apparatus. A profound sensorineural component of hearing loss usually is not present.1 In general, anomalies of the external and middle ear are more common than those of the inner ear. The incidence of EAC atresia is one in 10,000 to 20,000 at birth and is bilateral in one third of cases, but it appears to be more common in the United States than elsewhere.2 Imaging: A spectrum of involvement is seen, with only mild bony stenosis of the EAC present in some children. When the canal is atretic, the atresia may be membranous or bony. The severity of middle ear anomalies correlates with the extent of external malformation. Preoperative high-resolution computed tomography (CT) plays an integral role in determining potential candidates for hearing restorative surgery and should be performed shortly before intended surgery.3 If a sensorineural component to the hearing loss is present, magnetic resonance imaging (MRI) may offer additional information that may affect treatment. Ossicular anomalies often are found in association with EAC atresia. The handle of the malleus may be fused to the atretic plate (e-Fig. 10-1). Less commonly, the malleus may be fused to the incus, with severe dysplasia usually present. The incudostapedial joint can be involved as well and can be well demonstrated on axial and coronal images. Patent oval and round windows are essential in achieving a good surgical outcome. Atresia of the oval window often is seen with syndromic EAC atresia (Fig. 10-2). The stapes can be dysplastic, malpositioned, or absent. Significant abnormalities of the stapes often necessitate removal and placement of a prosthesis. The size of the middle ear cavity is significant, because small hypoplastic cavities are associated with a poor surgical outcome. The degree of mastoid pneumatization also may affect the surgical approach.3–5 Figure 10-2 A 5-year-old with bilateral microtia and conductive hearing loss. e-Figure 10-1 A 10-year-old with left conductive hearing loss and microtia. The course of the facial nerve is critical to the evaluation of children with EAC atresia. The most common anomaly involves anterior displacement of the descending segment of the facial nerve, with the stylomastoid foramen lying just posterior to the temporal mandibular joint. The posterior genu usually is more anteriorly positioned immediately posterior to the level of the oval window6 (Fig. 10-3). Less commonly, the tympanic segment may be malpositioned into the oval window niche or even more inferiorly over the cochlear promontory. The anomalous position of the tympanic segment of the facial nerve usually is associated with atresia or stenosis of the oval window. The facial nerve can be hypoplastic and difficult to visualize, but other labyrinthine segment anomalies are uncommon.7 Figure 10-3 External auditory canal atresia. Syndromic EAC atresia usually is associated with more severe middle ear anomalies and a higher incidence of inner ear anomalies; these patients are poorer surgical candidates. Syndromes associated with EAC atresia include Goldenhar syndrome, Treacher Collins syndrome, and Pierre Robin sequence8,9 (Tables 10-1 to 10-4; e-Fig. 10-4). Table 10-1 Otocraniofacial Syndromes with Congenital Abnormalities of the Ear EAC, External auditory canal; LSCC, lateral semicircular canal. From Hasso AN, Casselman JW, Broadwell RA. Temporal bone congenital anomalies. In Som PM, Carter HD, editors: Head and neck imaging. 3rd ed. St Louis: Mosby; 1996:1352-1355. Table 10-2 Otocervical Syndromes with Congenital Abnormalities of the Ear EAC, External auditory canal; IAC, internal auditory canal. From Hasso AN, Casselman JW, Broadwell RA. Temporal bone congenital anomalies. In Som PM, Carter HD, editors: Head and neck imaging. 3rd ed. St Louis: Mosby; 1996:1352-1355. Table 10-3 Otoskeletal Syndromes with Congenital Abnormalities of the Ear EAC, External auditory canal; IAC, internal auditory canal. From Hasso AN, Casselman JW, Broadwell RA. Temporal bone congenital anomalies. In Som PM, Carter HD, editors: Head and neck imaging. 3rd ed. St Louis: Mosby; 1996:1352-1355. Table 10-4 Miscellaneous Syndromes with Congenital Abnormalities of the Ear EAC, External auditory canal; LSCC, lateral semicircular canal. From Hasso AN, Casselman JW, Broadwell RA. Temporal bone congenital anomalies. In Som PM, Carter HD, editors: Head and neck imaging. 3rd ed. St Louis: Mosby; 1996: 1352-1355. e-Figure 10-4 A 4-year-old with Treacher Collins syndrome. Treatment: Grading systems have been developed to assess the probability of surgical success. These systems take into account such findings as middle ear volume, ossicular anomalies, and the status of the oval window.2 Transmastoid and anterior surgical approaches are used, with formation of a new tympanic membrane, mobilization of the ossicles, and possibly placement of a stapes prosthesis. The presence of a cholesteatoma also is a potential issue in both the imaging evaluation and surgical management of these patients. Clinical Presentation and Etiologies: Minor ear anomalies are associated with a normal external ear canal and are much less common than EAC atresia. Children with minor ear anomalies usually present with a nonprogressive conductive hearing loss. Ossicular abnormalities may be unilateral or bilateral. Unilateral ossicular abnormalities are usually sporadic and isolated. Bilateral anomalies may be associated with autosomal-dominant inheritance, and syndromic associations include Goldenhar and Treacher Collins syndrome. Round window atresia is caused by a deficiency in the development of cartilage during ossification of the otic capsule and typically is associated with conductive hearing loss. Oval window atresia is likely caused by anomalous development of the second branchial arch (innervated by the seventh cranial nerve) and often is associated with a mixed sensorineural conductive hearing loss. Oval window atresia can be seen with the CHARGE association (Coloboma of the eye, Heart defect, Atresia of choanae, Retarded growth and development, Genital anomalies, and Ear defect with deafness). Imaging: High-resolution CT using both axial and reconstructed coronal planes is the imaging modality of choice in evaluating patients with isolated middle ear anomalies. Anomalies of the stapes include aplasia, partial absence, columnar deformity, fusion of the head to the promontory, and footplate fixation. The incus can be aplastic, it can be fused to the lateral semicircular canal or scutum, it can have a malformed long process, it may be absent, or it may be fixed to an incudostapedial joint (e-Fig. 10-5). Similar anomalies involve the malleus; rarely, a bony bar fixes the malleus to the anterior lateral epitympanic wall9,10 (Fig. 10-6). These anomalies have been classified into five classes on the basis of their surgical success rate and include (from better to worse): Figure 10-6 A 10-year-old boy with right-sided conductive hearing loss. e-Figure 10-5 A 6-year-old with right-sided conductive hearing loss and no history of middle ear disease or surgery. Class 1: Normal stapes with abnormal incus or malleus Class 2: Mobile stapes footplate Class 3: Stapes footplate fixation only Class 4: Stapes footplate fixation with other anomaly Congenital stapes deformity or fixation may be associated with a perilymphatic fistula.13 Round window atresia is not associated with malposition of the facial nerve or ossicular abnormalities, but it has been reported in association with oval window atresia.14,15 Atresia of the oval window can occur in the absence of EAC atresia or stenosis. The absent oval window often is associated with malposition of the tympanic segment of the facial nerve into or inferior to the atretic oval window. The stapes typically is deformed or hypoplastic and appears oriented toward a segment of facial nerve rather than the oval window. The long and lenticular portions of the incus also are malformed. Anomalous development of the second branchial arch is likely the cause, with the facial nerve, incus, and a portion of the stapes arising from this structure.16,17 (Fig. 10-7). Figure 10-7 A 7-year-old with mixed hearing loss and a 60-dB threshold. The CHARGE association has typical features on high-resolution CT of the temporal bone, and oval window atresia is present in nearly all cases (e-Fig. 10-8). Anomalies of the incus and stapes are found along with fixation of the malleus to the anterior tympanic wall. The inner ear is malformed in all cases, with the cochlea and vestibular system involved. The typical vestibular findings are a small vestibule and absent semicircular canals. The facial nerve often is difficult to identify in these patients and may be hypoplastic or malpositioned.18,19 e-Figure 10-8 A 9-year-old with CHARGE syndrome. Treatment: Ossicular chain reconstruction is the mainstay of treatment. Accurate preoperative assessment is essential to plan the appropriate procedure. The status of the oval window is important because atresia imparts a poor surgical prognosis. Determining the position of the facial nerve is essential in these patients. Clinical Presentation and Etiologies: Study of the genetic defects associated with congenital sensorineural hearing loss (SNHL) is increasing. Nonsyndromic congenital hearing loss is not associated with additional medical anomalies and constitutes 70% of the cases. Genetic mutations are found in persons with nonsyndromic congenital hearing loss, with approximately 50% of nonsyndromic recessive hearing loss caused by a mutation in the GJB2 gene.20 Children with enlarged endolymphatic ducts have mutations of the SLC26A4 (PDS) gene in up to 82% of the hereditary cases and 30% of the sporadic cases.21 Auditory neuropathy/dyssynchrony may be associated with mutations in the OTOF gene, as well as suggested by the associated brain anomalies. These patients typically have a poor response to hearing aids and may benefit from earlier cochlear implantation.22,23 The future classification of congenital hearing loss is likely to be based on a combined genetic and morphologic evaluation. Syndromic congenital hearing loss accounts for 30% of cases. A wide spectrum of syndromes is associated with congenital anomalies of the ear, which include inner ear malformations (Tables 10-1 through 10-4).9 Toriello et al.24 also have produced a definitive text on syndromic associations and congenital hearing loss. It is important to understand that diagnosing congenital SNHL can be difficult in sporadic cases, and perinatal insults can be responsible. Congenital cytomegalovirus infection, hypoxia, ototoxic drugs, kernicterus, and, rarely, tumors are additional etiologies that should be considered and are at times suggested by imaging25 (Fig. 10-9). Figure 10-9 Congenital sensorineural hearing loss.
Congenital and Neonatal Abnormalities
External Auditory Canal Atresia
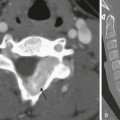
A coronal high-resolution computed tomography image at the expected level of the oval window demonstrates that the middle ear cavity is markedly hypoplastic and opacified (curved arrow). The oval window is atretic and the facial nerve is not well demonstrated (straight arrow). This patient would be a poor surgical candidate for middle ear reconstructive surgery.
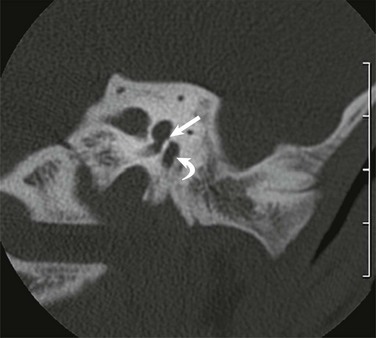
A, Coronal high-resolution computed tomography shows bony atresia of the external auditory canal and fusion of the malleus to the atretic plate (arrow). B, The stylomastoid foramen is positioned anteriorly (arrow).
A coronal high-resolution computed tomography image shows anterior displacement of the descending facial nerve (arrow) with the posterior genu at the level of the oval window.

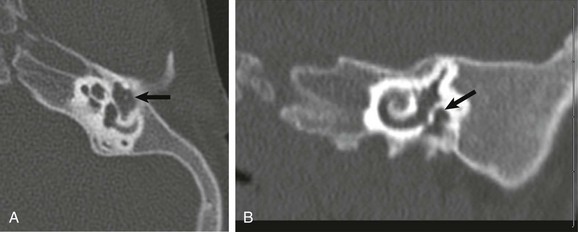
A and B, Axial and coronal high-resolution computed tomography images show a markedly hypoplastic middle ear cavity with absent ossicles (arrows). The external auditory canal and oval window are atretic.
Isolated Middle Ear Anomalies
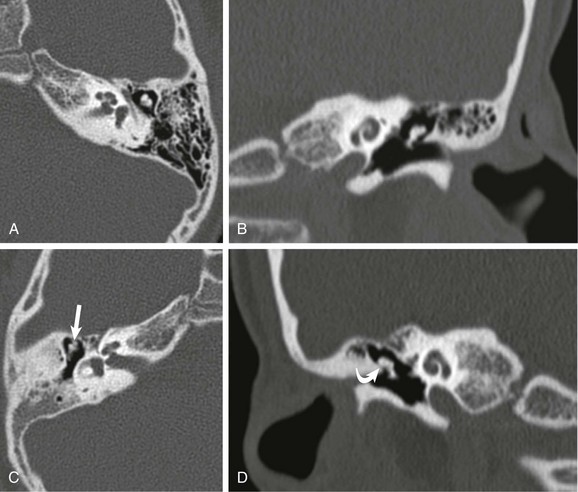
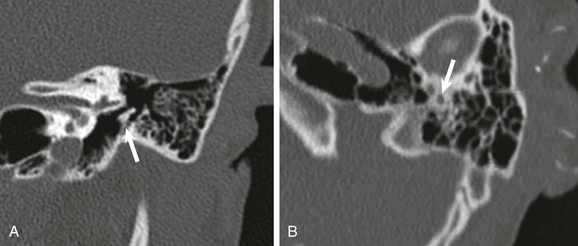
A and B, Axial and coronal high-resolution computed tomography through the normal left middle ear. C and D, Corresponding axial and coronal computed tomography images of the right middle ear shows incomplete separation of the malleolar head from the tegmen (straight arrow) and narrowing of Prussak space (curved arrow). Ossicular fixation was confirmed surgically.
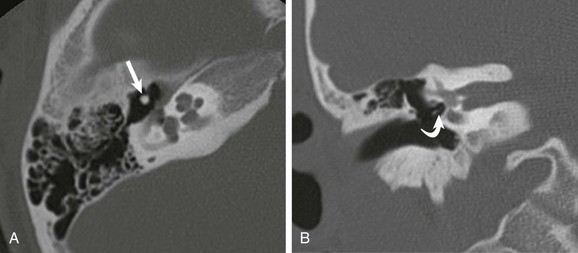
A and B, Axial and coronal high-resolution computed tomography images through the middle ear. Congenital absence of the short and long process of the incus is noted. The head of the malleus (straight arrow) and lenticular process of the incus (curved arrow) are present.
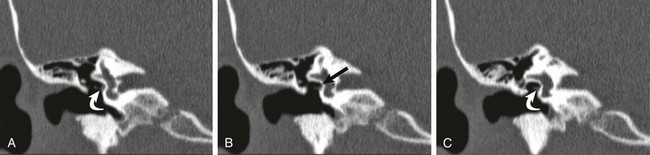
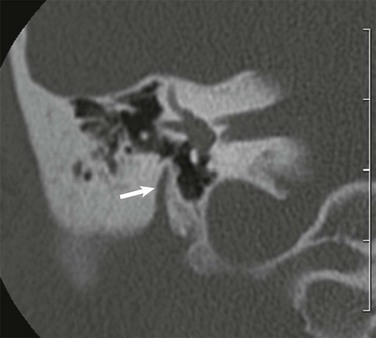
A, B, and C, Coronal high-resolution computed tomography images from anterior to posterior. The oval window is atretic (arrow), with displacement of the facial nerve within and slightly below the structure (curved arrows). The stapes was not visualized, and the lenticular process of the incus appears adherent to the displaced facial nerve.
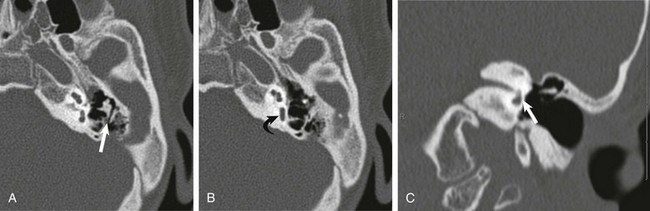
A and B, Axial high-resolution computed tomography (CT) images demonstrate a very hypoplastic vestibule (curved arrow) and absent semicircular canals. The short process of the incus is fixed posteriorly (straight arrow) and the sinus tympani is absent. C, A coronal CT image shows absence of the oval window (arrow).
Inner Ear Anomalies
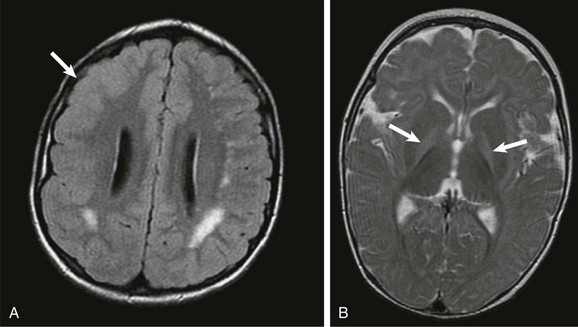
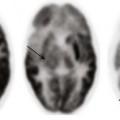
A, An axial fluid attenuated inversion recovery image shows that an abnormal increased T2-weighted signal is present within the supratentorial white matter. Cortical dysplasia is present within the right frontal region (arrow). The findings are consistent with congenital cytomegalovirus infection. B, An axial T2-weighted image through the basal ganglia in a young child with sensorineural hearing loss and a history of prematurity shows an increased T2-weighted signal in the bilateral globus pallidus (arrows), consistent with kernicterus.