Congenital Anomalies of the Spine and Spinal Cord: Embryology and Malformations
Thomas P. Naidich
Andres W. Su
Bradley N. Delman
David G. McLone
Robert A. Zimmerman
Susan I. Blaser
Charles A. Raybaud
Nolan R. Altman
Asim F. Choudhri
In the spine, the most common congenital lesions presenting to medical attention are the diverse forms of spinal dysraphism and the diverse forms of caudal spinal anomalies (1). These conditions are typically discovered at birth or in early childhood. Less often, they first present to medical attention in adulthood (2,3,4). The widespread use of magnetic resonance imaging (MRI) has reduced the incidence of delayed diagnosis of these conditions and disclosed surprising numbers of adult dysraphics.
Congenital malformations of the spine are grouped into three broad categories (1,5,6). The first category is spina bifida aperta, which comprises those lesions in which the neural tissue and/or meninges are exposed to view in the midline of the back. The most common forms of spina bifida aperta are the myelocele and myelomeningocele. Simple meningoceles without skin cover are less common. The second category is occult spinal dysraphism, which comprises those lesions in which the malformed neural tissue lies deep to an intact skin cover (1,7). This is a heterogeneous group of lesions that includes dorsal dermal sinus, spinal lipoma, tight filum terminale syndrome, neurenteric cyst (NEC), and diastematomyelia. These lesions share the common features that the spinal cord is cleft and/or is tethered in an abnormally low position by a fibrous band, a bone spur, or a caudal mass (8). The specific cause of the tethering may often be unknown (9). Midline cutaneous stigmata such as skin dimples, skin tags, hemangiomatous nevi, and patches of hypertrichosis frequently overlie the zone of abnormal nervous tissue and signal the presence of “hidden” pathology. The third category is caudal spinal anomalies, which comprises those lesions in which malformations of the distal end of the spine, spinal cord, and meninges are associated with disorders of the hindgut, kidneys, urinary bladder, and genitalia. These include sacral agenesis, terminal myelocystocele, and anterior sacral meningocele, among others.
This chapter addresses the most common forms of dysraphism and the most common caudal spinal anomalies (10,11). It provides an overview of embryology for orientation and then groups the anomalies in terms of the specific derangements of embryology that are believed to give rise to the anomalies (12,13). More detailed discussions of embryogenesis may be found in the excellent texts by Bruce M. Carlson (14) and Scott F. Gilbert (15). It is hoped that this approach will provide greater understanding of these lesions and better long-term retention of information about them. A conceptual framework of embryology and pathogenesis may eliminate the need for rote memorization; certainly it is a more satisfying approach intellectually (1,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31).
Overview of Normal Embryogenesis
Early Events
Two-Layered Germinal Disc
Toward the end of the second week of gestation, the embryo has taken the form of a two-layer disk composed of a superficial layer of columnar epithelium that faces the amniotic cavity and a subjacent layer of cuboidal cells that faces the yolk sac (future gut) (Fig. 19.1). The superficial layer is designated the epiblast. This rests on a basal lamina (14). The subjacent layer is designated the hypoblast (32). The epiblastic cells will give rise to (nearly) all future ectoderm, mesoderm, and endoderm of the embryo (32,33). The hypoblastic cells will ultimately form the extra-embryonic tissues (32).
Gastrulation Forms the Three-Layered Germinal Disc
By about day 13, signals from cells at the edge of the two-layered disc induce caudal epiblastic cells to condense into a linear primitive streak in the midline posteriorly (Fig. 19.1) (32) (14). This streak identifies the craniocaudal and left–right axis of the embryo (10,11,12,13,33,34). The primitive streak exhibits a midline primitive groove. On each side of the primitive groove, epiblastic epithelial cells reduce their expression of the epithelial cell adhesion molecules (E-CAMs), separate from each other, detach from the basal lamina, and become individual mesenchymal cells. This process is designated epithelial–mesenchymal transition. The newly mesenchymal cells then migrate medially into and through the primitive groove to establish the three future cell layers of the embryo.
Cells traversing different portions of the primitive groove form distinct lineages as they emerge from the groove (14). The first migratory epiblastic cells pass inward through the posterior portion of the primitive groove. These cells displace nearly all of the hypoblastic cells laterally and become nearly all of the endoderm of the embryo (32). The next wave of migratory epiblastic cells enters the primitive groove further anteriorly and then passes laterally, deep to the epiblast, to form the mesodermal layer (Fig. 19.2A). These newly mesodermal cells then migrate outward from the midline along defined routes to establish the paraxial, lateral, and cardiac mesoderm (14). The last wave of epiblastic cells migrates inward through the anteriormost portion of the primitive groove to establish the midline axial structures, including the primitive node
(synonym: Hensen’s node), the prechordal plate, and the notochord (14,15). The epiblastic cells that remain on the surface form the surface ectoderm. By these processes, the bilaminar disk composed of epiblast and hypoblast converts into the trilaminar disk formed by endoderm, mesoderm, and ectoderm (32). The process by which cells migrate to the primitive streak, ingress, and establish new cell layers is designated gastrulation.
(synonym: Hensen’s node), the prechordal plate, and the notochord (14,15). The epiblastic cells that remain on the surface form the surface ectoderm. By these processes, the bilaminar disk composed of epiblast and hypoblast converts into the trilaminar disk formed by endoderm, mesoderm, and ectoderm (32). The process by which cells migrate to the primitive streak, ingress, and establish new cell layers is designated gastrulation.
 FIGURE 19.1 Bilaminar disk. Prochordal plate. A: Longitudinal section. B: A View of the epiblastic surface that faces the amnion. By the end of the second week of gestation, the two-layered embryo consists of an epiblastic layer (c) that faces the amnionic cavity (1) and an hypoblastic layer (en) that faces the yolk (vitelline) sac (2). The prochordal plate (P) establishes the cephalic end of the bilaminar disk and the first craniocaudal left-right axes of the embryo. The primitive streak (arrowhead) forms in the midline toward the caudal end of the embryo. The thickening at the cephalic end of the streak, designated Hensen’s node, contains a small primitive pit. (From Raybaud CA, Naidich TP, McLone DG. Développement de la moelle et du rachis. In: Manelfe C, ed. Imagerie du rachis et de la moelle. Paris: Editions Vigot, 1989, with permission.) |
 FIGURE 19.2 Trilaminar disk. Migrations. A: Longitudinal section. B: View of the epiblastic surface that faces the amnion. C: Transverse section through the notochord at a slightly later time when the notochord has begun to induce formation of neural ectoderm (NE). Epiblastic cells pass medially to enter the primitive streak, descend to the interlaminar level, and then migrate cephalically and laterally between the ectoderm (c) and the endoderm (en). This migration converts the bilaminar disk to the trilaminar disk. Those cells that migrate in the true midline will form the notochordal process (N). The primitive pit will deepen and invaginate into this process to form the notochordal canal. Cells that migrate in the paramedian location will form the paraxial mesoderm (M), which becomes somites. Cells that migrate in the intermediate and the lateral locations will form the intermediate and lateral mesenchyme. (From Raybaud CA, Naidich TP, McLone DG. Développement de la moelle et du rachis. In: Manelfe C, ed. Imagerie du rachis et de la moelle. Paris: Editions Vigot, 1989, with permission.) |
Primitive (Hensen’s) Node, Prechordal Plate, and Notochordal Process
Epiblastic cells that migrate to the anterior most end of the primitive streak help to form the primitive node (Hensen’s node) (14). The node contains a primitive pit. The first epiblastic cells to pass through the primitive node form a discrete midline cell mass designated the prechordal plate (Fig. 19.2B) (14). The prechordal plate helps to establish and maintain the signaling needed for the future head (14). Cells from the node, and the next group of epiblastic cells to traverse the node, form a stream of mesendodermal tissue that ultimately develops into the definitive notochord in the midline directly between the endoderm and the ectoderm. The notochord directs the differentiation of the overlying ectoderm into specialized neural ectoderm (Fig. 19.2C). The notochord will also direct the organization of the future vertebral centra and disks (35,36). From these simple beginnings, subsequent steps will form the cephalic portion of the spinal cord and then the caudal (i.e., distal) cord (10,11,12,13).
The cephalic portion of the spinal cord forms by the mechanism of primary neurulation. Primary neurulation is an orderly sequence of steps in which the neural ectoderm forms a neural plate that then folds up into a neural tube. This mechanism establishes the cervical, the thoracic, and some of the lumbar portions of the spinal cord. A smaller, distalmost portion of the lumbar cord and the filum terminale form by a separate sequence of agglomeration of cells into a cell mass, vacuolation of that cell mass, and partial involution of the tissue formed. This process is designated secondary neurulation. Recent evidence from chick embryos suggests that—at least in chicks—the upper and lower portions of the spinal cord made by primary and secondary neurulation may link together by a separate intermediate process designated junctional neurulation (37). Because these different portions of the spinal cord form by distinctly different mechanisms, they exhibit distinctly different types of malformation.
Normal Neurulation
Figures 19.3 and 19.4 provide an overview of normal neurulation. Figures 19.5 and 19.6 add additional detail (38,39).
Convergent Extension
By the end of the third week, the notochord directs the overlying ectoderm toward a euroectodermal identity. The neural plate is initially slipper-shaped and rests on a basement membrane (38,39). It is one cell layer thick. The slipper-shaped neural plate then elongates rostrocaudally, narrows transversely, and thickens (apicobasally), except in the true midline (38). Because the future spinal tissue narrows from side to side as it elongates rostrocaudally, this process is designated convergent extension (Fig. 19.5).
Forming the Neural Folds
The developing neural plate is then folded into a neural tube by flexing the neural plate at midventral and paired dorsolateral “hinge points” (Figs. 19.3 and 19.6). Each cell in the single cell layer of the neural plate faces two distinct environments. One surface of the cell faces the amnion (and future lumen of the neural tube). This surface is designated the apical surface. The opposite (abluminal) surface of the cell is anchored to the basement membrane. This surface is designated the basal surface. In mammalian cells, a “fence” of tight junction complexes partially separates the apical from the basal portion of each cell (40). Protein complexes within the apical and basal “compartments” are mutually antagonistic, distribute within the cell in an asymmetric, polarized fashion, and thereby “polarize” the cell (41). This difference in internal structure is designated apicobasal polarity or planar cell polarity (40).
Adherens junctions located toward the apical side of each cell link to ropes of actomyosin that run mediolaterally from cell to cell along the apical aspect of the neural plate. During neurulation, these links yoke the cells together to maintain cell cohesion as the neural plate raises up neural folds and ultimately closes into a neural tube (40).
The position of the nucleus within the cell plays an important role in neurulation, because the large cell nucleus occupies most of the total cell volume (40). At the future hinge points, the nuclei of adjacent cells position themselves along the basal (abluminal) surface of each cell. This localized positioning of the large nuclei away from the lumen expands the basal dimension of each cell and narrows the apical dimension of each cell, helping to flex the neural plate at the future hinge points (Fig. 19.6). Extrinsic forces such as the growth pressure of adjacent tissue also act to elevate and bend the neural tube. In experimental settings, isolated neural plate will shape and furrow normally but will not fold. Conversely, after extirpation of mesoderm, endoderm, and associated extracellular matrix (but not epidermal ectoderm), the neural folds will elevate, converge, and often even fuse (38).
Fusion of the Neural Folds
As the neural folds come together, the neural and epidermal ectoderm meet in the midline dorsally, adhere, and fuse together. Fold adherence, and later fold fusion, involve the synthesis of complex cell surface glycoproteins (glycosaminoglycans) and elaboration of localized protrusions (filopodia) from the neuroepithelial cell membranes, especially on their luminal surfaces (32). The filopodia may help to align the converging neural folds or help to pull the apposing surfaces together. Successful closure of the neural tube also involves folic acid and folic acid receptors at the surface of the closing folds (42). The precise mechanics of adherence and fusion appear to vary with the specific species studied and with the specific level along the neural axis.
Neural Crest Migration
Neural crest cells arise in the lateral tips of the rising neural folds and then migrate widely to become all of the glia and most of the neurons of the peripheral nervous system; distinct endocrine and paraendocrine cells, the chromaffin cells of the adrenal medulla; all of the melanocytes of the body (except for the retinal pigment); and most of the skeletal and connective tissues of the face, head, and neck (43). The premigratory neural crest cells undergo epithelial–mesenchymal transition, delaminate from the dorsal neural ectoderm, and then migrate to their final destinations (43,44). The precise details of the time at which the neural crest cells detach; whether they detach from the elevating folds, from the converging folds, or from the incipient roof plate; and their specific paths of migration vary across species and across spinal levels within each species (32,38).
Points of Closure of the Neural Tube
The neural folds are believed to meet first and fuse in the region of the caudal rhombencephalon (future occipital region) at about postovulatory day 22 (13,32,45). Some experimental data in mice and study of human malformations have raised the possibility that the mammalian neural tube closes independently at multiple sites, perhaps five (46). The multiple closures proposed would include the traditional first closure between somites 2 and 4 that “zips” caudally to form the cervical–thoracic
cord down to a posterior neuropore at L2 and an additional closure distal to L2 that extends caudally to form the segment of neurulated spinal cord between L2 and S2.
cord down to a posterior neuropore at L2 and an additional closure distal to L2 that extends caudally to form the segment of neurulated spinal cord between L2 and S2.
 FIGURE 19.3 Three stages of neurulation. Axial sections of mammalian embryos oriented like axial magnetic resonance imaging with ventral toward the top. A: Neural plate. The primitive central nervous system originates as a “flat” plate of neural ectoderm (NE) composed of a single layer of cells with two distinct surfaces. The ventral surface of the neural plate faces the interior of the embryo. With folding, the ventral surface will form the entire external surface of the spinal cord. This ventral surface is contiguous with the notochord (N) in the midline and with the loose mesenchyme (M) to each side. The dorsal surface of the neural plate faces the exterior of the embryo and the amniotic cavity (open arrows indicate the amnion). With folding, the dorsal surface will form the inner surface of the spinal cord around the central canal of the cord. This dorsal surface exhibits a midline sulcus or furrow (black arrow) called the ventral neural groove (median longitudinal furrow) because it will become the ventral groove of the central canal of the cord. The epidermal ectoderm (large curved arrows) forms a thin layer of cells that encloses the embryo ventrally and laterally. This epidermal ectoderm is tightly adherent (small curved arrows) to the neural ectoderm at the lateral edges of the neural plate, precisely at the junction of the ventral and dorsal surfaces of the neural plate. At this stage the mesenchyme is restricted to two paramedian zones ventral to the neural plate: Mesenchyme is excluded from the midline by the contiguity between the primitive gut (G), the notochord (N), and the ventral surface of the neural epithelium. Mesenchyme is prevented from passing posterolaterally by the tight adherence of the epidermal ectoderm with the neural ectoderm at the margins of the neural plate. No mesenchyme can pass around to the dorsal aspect of the neural epithelium. B: Neural folds. With further development, the neural plate begins to elevate its lateral portion dorsally, creating the neural folds. The ventral surface of the neural epithelium is beginning to assume the appearance of the external surface of the future spinal cord. The future central canal appears between the folds as a dorsal channel with a ventral neural groove (vertical black arrow) and two lateral sulci called the sulci limitans (horizontal black arrows). These groves may also be designated the ventral hinge point and the paired dorsolateral hinge points, because the neural plate folds inward along these grooves. The epidermal ectoderm (large curved arrows) remains tightly adherent to the neural ectoderm (NE) precisely at the neural ridge (small curved arrows) and passes dorsally with the ridge as the neural plate folds. This tight adherence of epidermal with neural ectoderm prevents mesenchyme from migrating to the dorsal surface of the neural epithelium. The primitive gut remains contiguous with the notochord and the notochord with the ventral surface of the neural plate, excluding mesenchyme from the midline. The mesenchyme remains restricted to the two paramedian zones ventral to the developing neural tissue. The superficial membranes external to epidermal and neural ectoderm are the amnion. C: Neural tube. The neural folds come into apposition and fuse together, closing the neural tube. The old dorsal surface of the neural plate has now become the wall of the primitive central canal. The old ventral surface of the neural plate has now become the entire exterior of the neural tube. The epidermal ectoderm now lies in the dorsal midline but is still attached to the neural ectoderm. In a very carefully orchestrated series of events, the neural folds fuse, closing the neural tube. The epidermal ectoderm of each side separates from the neural ectoderm in a process called disjunction. The left and right layers of epidermal ectoderm also fuse in the midline dorsal (superficial) to the neural tube, enclosing the future central nervous system beneath the future skin. |
 FIGURE 19.4 Diagrammatic representation of neurulation. A,B: Summary of events illustrated in Figure 19.3A,B. The short lines at the apical ends of the neuroepithelial cells represent actin filaments, contraction of which may play a role in bending the neural plate into the neural folds. Curved arrows indicate epidermal ectoderm. C: After closure of the neural tube, the epidermal ectoderm disjoins from the neural ectoderm and fuses in the dorsal midline superficial to the neural tube. The notochord (N) and the primitive gut then migrate ventrally, separate from each other, and separate from the neural tube, permitting mesenchyme (M) to migrate around the notochord. The perichordal mesenchyme will be induced by the notochord to form the vertebral centra and intervertebral disks (2). The epidermal ectoderm separates more widely from the neural ectoderm, permitting the mesenchyme (M) to migrate dorsal to the neural tissue. This mesenchyme will be induced by the neural tube to make the neural arches and lateral elements. Mesenchyme condensing ventrolateral and dorsolateral to the developing vertebrae will become the paraspinal musculature. cc, central canal. (From Naidich TP, Gorey MT, Raybaud C, et al. Malformations congénitales de la moelle. In: Manelfe C, ed. Imagerie du rachis et de la moelle. Paris: Editions Vigot, 1989, with permission.) |
 FIGURE 19.5 Diagrammatic representation of the four stages of neurulation. In this schema, the dorsal surface of the embryo is displayed superiorly and the ventral surface inferiorly. A: Gross morphology. The neuroectodermal cells at the hinge points show a wedge shape. B: The intrinsic forces acting during flexion and elevation of the neural folds. C: The extrinsic forces acting in these processes. See text for explanation. (From Smith JL, Schoenwolf GC. Neurulation: coming to closure. Trends Neurosci 1997;20:510–517, with permission.) |
 FIGURE 19.6 Epithelial organization of the early neural plate and formation of hinge points. A: Cross-sectional (apicobasal) diagram of the single cell layer pseudostratified neural epithelium and the changing position of the nucleus during interkinetic nuclear migration. The large nuclei attach to both the apical surface of the cell (red) and the basal surface (blue). During the cell cycle (M, G1, S, G2, M) the nucleus migrates between the apical and basal surfaces of the cell, but undergoes cell division near the apical surface. B: Apicobasal cell polarity. The apical (red) surface of the cell faces the future lumen. The basolateral (green) surface is related to the basement membrane (BM). The apical and basal compartments of each cell are separated by tight junctions (TJ). Adherens junctions (AJ) are located on the basal side of the tight junctions and are associated with the adherens/actin belt (gray). The adherens and tight junctions are associated with an apical polarity complex (PAR) that antagonizes and excludes the basolateral proteins such as LGL (lethal giant larva). C: Away from the hinge point, cells may display asynchronous cell cycle progression, with the nuclei distributed variously along the apicobasal axis. D: At the median hinge point, however, the majority of the cells are situated basally, in the same phase of the cell cycle. Although the number of cells is equal in C and D, the positioning of the cell nuclei contributes to an apical narrowing and basal widening of the epithelium. (Redrawn after Eom DS, Amarnath S, Agarwala S. Apicobasal polarity and neural tube closure. Dev Growth Differ 2013;55:164–172.) |
Disjunction of Neural from Epidermal Ectoderm
After the neural folds fuse into the neural tube, the epidermal ectoderm of each side separates from the neural ectoderm in a process designated disjunction. Differential expression of the neural cell adhesion molecule (N-CAM) and the epidermal cell adhesion molecule (E-cadherin, previously designated E-CAM) confers different adhesion properties on each cell type, allowing the cutaneous ectoderm to disjoin from the neural ectoderm. The left and right portions of the disjoined superficial (epidermal) ectoderm then fuse together dorsal to the neural tube, to close the superficial ectoderm (future skin) in the midline (Fig. 19.4C).
Following disjunction, portions of the subjacent mesenchyme migrate ventrally around the notochord to establish the future vertebral centra and discs. Other portions of the mesenchyme migrate dorsally around the neural tube, burying it beneath a thick layer that ultimately forms the meninges, neural arches, and paraspinal muscles (Fig. 19.4).
Molecular Signaling
Neurulation, neural crest migration, specification of cell identity, and patterning along the body axes are all orchestrated by cascades of molecular signals including the ventralizing signal sonic hedgehog (SHH, Shh) and the dorsalizing signals: bone morphogenic protein (BMP) and wingless genes (Wnts) (Figs. 19.7 and 19.8). During late gastrulation and early neurulation, Hensen’s node (the “organizer”) causes the notochord to secrete the signaling molecule SHH. SHH induces the neuroepithelium of the ventral hinge point to transform into a floor plate that then produces additional SHH. The SHH diffuses outward from the notochord and floor plate to establish a precise and progressively diminishing gradient of signal. The precise intensity of signal along that gradient modulates the expression of paired box (Pax) genes (Fig. 19.8) and ultimately defines specific cell compartments in which the ventral neuroectodermal cells become motor neurons or distinct types of ventral interneurons (Fig. 19.9A) (38,47,48,49). SHH also induces the ventral somatic mesoderm to become sclerotome (38,47,48).
In similar fashion, BMPs and Wnts produced by the epidermal ectoderm and mesoderm during and after neurulation diffuse outward to establish a precise and progressively diminishing gradient of BMP and Wnt signal. The precise intensity of signal along that gradient modulates the expression of different paired box (Pax) genes (Fig. 19.8) to establish dorsal compartments in which dorsal neuroectodermal cells assume specific dorsal fates (Fig. 19.9A) (49). The BMPs and Wnts also induce the adjacent portions of the somatic mesoderm to become dermomyotomes (see later Fig. 19.104) (38,47,48). After the cell compartments are established and the neurons assume their specific fates, the relative sizes of each compartment and the numbers of cells within each compartment are determined by
modulating the rates of proliferation and differentiation of the cells in that compartment (Fig. 19.9B) (50).
modulating the rates of proliferation and differentiation of the cells in that compartment (Fig. 19.9B) (50).
 FIGURE 19.7 Inductive signals during spinal cord development. The dorsal surface is displayed superiorly. A: Gross stages in the development of the embryonic spinal cord. The neural plate is generated as a columnar epithelium. The axial mesodermal cells of the notochord (N) and the paraxial mesodermal cells (future somites) (S) both lie ventral to the neural plate. Epidermal ectoderm (ECT) flanks the plate laterally. The floor plate develops over the notochord in the midline. Fusion of the dorsal tips of the neural folds forms the neural tube, generates roof plate cells in the dorsal midline, and leads to the emigration of neural crest cells (NC). Neuroepithelial cells proliferate and differentiate into neurons located at different dorsoventral positions. Subclasses of motor neurons and ventral interneurons differentiate ventrally, near the floor plate, whereas commissural (C) and association (A) neurons differentiate dorsally, close to the roof plate. Dorsal root ganglion (DRG) neurons differentiate from postmigratory neural crest cells. B: Diagrammatic representation of the inductive signals. Sonic hedgehog (SHH; blue) is associated with ventral cell fates (ventralizing influence). BMPs (orange) are associated with dorsal cell fates (dorsalizing influences). SHH is initially expressed in the axial mesoderm. BMPs originate in the epidermal ectoderm flanking the lateral edges of the neural plate. At neural fold stages, SHH becomes expressed by floor plate cells in the ventral midline of the developing cord, whereas BMPs are expressed by cells in the dorsal tips of the neural folds. After closure of the neural tube, BMP expression is lost from the epidermal ectoderm except in the dorsal midline but now appears in the roof plate and the adjacent neural tube. At the onset of neuronal differentiation, SHH expression is maintained by the floor plate, whereas BMP expression persists in the dorsal neural tube. C: Pax gene expression during neurulation. In chick embryos, the regulated expression of three Pax genes (Pax3, Pax6, and Pax7) subdivides the developing neural tube into distinct domains. Caudal neural plate cells at all mediolateral positions initially express Pax3 and Pax7. At neural fold stages, Pax3 and Pax7 expression is repressed medially, whereas Pax6 expression is detected at all mediolateral positions except in the midline. After neural tube closure, Pax3 and Pax7 expression becomes restricted to the dorsal half of the neural tube, whereas Pax6 is expressed by more-ventral cells and by cells in the dorsal half of the neural tube. The expression of Pax6 is correlated with somitogenesis and is progressively upregulated in the neuroepithelium of each newly formed somite (376). N, notochord; FP, floor plate; R, roof plate. (Those interested in the later stages of neuronal differentiation will find additional information and diagrams later in this chapter.) (From Tanabe Y, Jessell TM. Diversity and pattern in the developing spinal cord. Science 1996;274:1115–1123, with permission.) |
Patterning the Body Axes
Rostrocaudal Patterning
Rostrocaudal patterning of the spine and the spinal cord is controlled by the precisely orchestrated and sequential expression of both maternal genes and embryonic genes (14). Homeotic genes (synonyms: homeobox genes, Hox genes) determine the specific identity of the individual segments of neural tissue, neural crest, paraxial mesoderm, and surface ectoderm. They specify the features that are characteristic of the first cervical segment (C1), for example, rather than those characteristics of the first thoracic segment (T1) or lumbar segment (L1) (Fig. 19.10) (14,15). The Hox genes appear to act through a gradient of retinoic acid (RA) concentration that increases progressively from cranial toward caudal. As the vertebrate embryo enters neurulation and starts to elongate, paired somites form sequentially on each side of the spinal cord. RA is produced by the somites and the anterior presomitic mesoderm in increasing concentration from cranial
to caudal, establishing the gradient of RA that specifies the precise identity of the individual mesodermal and neural segments (Figs. 19.11 and 19.12) (51).
to caudal, establishing the gradient of RA that specifies the precise identity of the individual mesodermal and neural segments (Figs. 19.11 and 19.12) (51).
 FIGURE 19.8 Distribution of Pax gene expression around the circumference of the neural tube. Note the positions of Pax3 and of Pax6. (From Carlson BM. Human Embryology and Developmental Biology. St. Louis, MO: Mosby; 1994, with permission.) |
Left–Right Asymmetry
Truncal asymmetry is induced by the asymmetric expression of signaling molecules on the future left and right sides of the embryo (Fig. 19.13) (38,52,53). At Hensen’s node, synchronous clockwise rotation of cilia induces a leftward flow of amnionic fluid across the node. This flow is sensed by perinodal crown cells. Differential activity of H+/K+-ATPase results in increased levels of extracellular Ca2+ on the left (54). This induces a regulatory cascade that directs left-side development (15,55). The precise mechanisms vary among vertebrate groups, but, overall, the asymmetrically greater levels of Ca2+ on the left stimulate Nodal-related genes to produce nodal protein on the left. Nodal protein activates the left-inducing transcription factor Pitx2 and represses a protein Snail related in the left-sided mesoderm (15). On the right, Fgf8 protein prevents expression of Nodal and Pitx2, preventing left-sidedness, and may also activate a specific right-determining program (15).
The initial expression of Nodal in the region about Hensen’s node is subsequently translated into a broad domain of Nodal expression in the left lateral plate mesoderm in a mechanism controlled by SHH (54). Interestingly, the symmetry-breaking steps that create the initial left–right asymmetry later interfere with the synchronous, symmetrical formation and union of paired somites (56). Transient, asymmetrical expression of RA is then required to buffer these effects and shield the presomitic mesoderm from the desynchronizing action of those early signals (56).
Deranged Primary Neurulation
Derangements of primary neurulation are often referred to as neural tube defects.
Spina Bifida Aperta: Myelocele and Myelomeningocele
The term spina bifida aperta designates those forms of spinal dysraphism in which the neural tissue and/or meninges are exposed to the environment, because the skin, fascia, muscle, and bone are deficient in the midline of the back. Myelocele and myelomeningocele are the two most common forms of spina bifida aperta.
 FIGURE 19.9 Patterning the closed neural tube. A: Differential induction of neuronal types. The opposing gradients of the ventralizing signal. (Reproduced with permission from Kicheva A, Bollenbach T, Ribeiro A, et al. Coordination of progenitor specification and growth in the mouse and chick spinal cord. Science 2014;345(6204):1254927.) B: Sonic hedgehog (Shh) emanating from the notochord (NC) and floor plate (FP) and the dorsalizing signals of bone morphogenic protein (BMP) and Wnts emanating from the epidermal ectoderm activate a cascade of signals that establish compartments in which cells take on specific neuronal identities. The motor neurons (MN) develop in a characteristic location in relation to the ventral interneurons V0–V3. The dorsal interneurons (dI1–dI6) develop in dorsal half of the cord. Sox2 confers neuronal identity on the progenitor (p) population that then migrates outward to its final location. |
 FIGURE 19.10 Rostrocaudal patterning by the Hox gene code. The symbols in the graph indicate the presence of activity of the Hox genes (upper horizontal axis) in each of the numbered vertebral segments (vertical axes) in the mouse and in the human. The individual identity of each segment of the vertebral axis is specified, in part, by the patterned expression of sequences of the homeotic genes (Hox genes). Expression of these Hox genes depends in turn on a rostrocaudal gradient of retinoic acid. (From Carlson BM. Human Embryology and Developmental Biology. St. Louis, MO: Mosby; 1994, with permission.) |
Before 1980, myelocele and myelomeningocele occurred in 1 to 2 per 1,000 live births, up to 8 per 1,000 live births in specific populations (57). Since then, the incidence of these malformations has been reduced sharply (up to 70%) simply by adding folate supplements to the diet of pregnant mothers in the period from before conception to 6 weeks after conception (58,59). As a result, the present incidence of myelocele and myelomeningocele in the United States is approximately 0.3 to 0.44 per 1,000 live births (60). The incidence may be higher in women with seizure disorders, because anticonvulsive medications such as phenobarbital, phenytoin, carbamazepine, and valproic acid lower the bioavailability of folate, leading to an increase in the incidence of neural tube defects in recipients (61). Such women require special care with folate supplementation.
The mechanism of action of folate is unclear. The folate levels in mothers of affected children are normal or only mildly depressed (62). Dietary folic acid uptake by these mothers is normal. Furthermore, folic acid deficiency does not directly cause neural tube defects in mice or cultured rat embryos (62). It may be that folic acid corrects a metabolic deficiency that would otherwise lead to neural tube defects, perhaps by preventing excessive incorporation of thymidine into DNA or perhaps by compensating for an inadequate supply of 5,10-methylene tetrahydrofolate in pyrimidine biosynthesis (62,63). Specific polymorphisms in the methylenetetrahydrofolate dehydrogenase (MTHFD1) gene, such as 1958G>A are known to be significantly associated with susceptibility to neural tube defects. Among these, homozygous AA carriers are more likely to suffer neural tube defects than are others with GA or GG genotypes (64). Neural tube defects are also known to be associated with disorders of maternal methionine metabolism and with elevated maternal levels of homocysteine. The greater availability of folate after folate supplementation may act to prevent methionine deficiency or homocysteine excess (32).
Because folate supplementation appears to correct only 70% of neural tube defects, other etiologies and treatments must also be sought (61,65). These include agents that interfere with cell mobility and contraction by disrupting the intracellular protein actin; calcium channel antagonists that hinder calcium-mediated microfilament contraction; and agents that disrupt the cell surface glycoprotein interactions needed to fuse the dorsal edges of the neural tube (66).
Neural tube defects have also been related to derangements in the paired box gene Pax3 that affects N-CAMs, complex basement membrane glycoproteins, and microfilaments in the apical region of the neuroepithelium (60). Pax3 mutations in the Splotch mouse and the corresponding human Pax3 mutation (Waardenburg syndrome I on chromosome 2q35-q37.3) are both associated with neural tube defects (Figs. 19.7C and 19.8) (62,67). Myelomeningocele may also be related to fragile X syndrome, a genetic disease caused by defects in the fragile X mental retardation gene (FMR-1) at Xq27.3 (68). Other chromosomal abnormalities such as full and partial aneuploidy are found in 4.4% to 17.3% of fetuses with spina bifida (69). Triploidy, trisomy 13, and trisomy 18 are among the most frequent of the full aneuploidal defects associated with spina bifida (69). Multiple diverse partial deletions and partial duplications of autosomes and sex chromosomes are also common (69).
 FIGURE 19.11 Rostrocaudal patterning of the axial spine. Compare with Figures 19.10 and 19.12. Homeotic transformation of the axial spine is induced by time-related and concentration-related gradients of retinoic acid. The vertebral levels are shown on the left. The transformation frequencies are indicated as percentages of the animals studied. The approximate boundaries of expression of parologous groups of Hox genes are shown on the right. (From Kessel M, Gruss P. Homeotic transformations of murine vertebrae and concomitant alterations of Hox codes induced by retinoic acid. Cell 1991;67:89–104, with permission.) |
 FIGURE 19.12 Homeotic transformation of rostrocaudal patterning. Newborn mouse specimens. The Alcian blue-alizarin red preparation stains cartilage blue and ossified calcium-containing tissue red. The variations in the vertebral pattern produced by exposure of mouse embryos to retinoic acid on day 7 or day 8 after timed matings ™ indicate that retinoic acid (RA) shifts the expression of the Hox code along a rostrocaudal axis. This shift respecifies segment identity, leading to transitional vertebrae and alterations in the number of costal elements. The normal mouse pattern is C7, T13, L6 (see also Figs. 19.10 and 19.11). A: Vertebral pattern C6, T13, L5 induced by RA at 7 days, 4 hours ™. B: Vertebral pattern C6, T13, L6 induced by RA at 7 days, 4 hours ™. C: Vertebral pattern C7, T14, L6 induced by RA at 8.5 days ™. D: Vertebral pattern C7, T5, L5 induced by RA at 8.5 days ™. E: Vertebral pattern C7, T13, L6 (wild-type, normal for mouse). (From Kessel M, Gruss P. Homeotic transformations of murine vertebrae and concomitant alterations of Hox codes induced by retinoic acid. Cell 1991;67:89–104, with permission.) |
 FIGURE 19.13 Molecular factors driving body asymmetry. Diagrammatic representation. The earliest signal appears shortly after gastrulation and is mediated by activin receptor II (axtRII) positioned along the right side of the primitive streak. This inductive signal is relayed to the lateral plate mesodermal cells as they migrate anteriorly from the primitive streak. The zinc finger protein Snail related (cSnR) also becomes expressed in lateral plate mesoderm on the right. A few hours later nodal (a member of the transforming growth factor β family) becomes selectively expressed in a broad stripe of lateral plate mesoderm on the left side of the body, roughly mirroring the distribution of cSnR on the right. The primitive heart tube then begins its program of looping and bends to the right, followed closely by a rightward rotation of the entire body axis. Shh, sonic hedgehog. (From Robertson EJ. Left-right asymmetry. Science 1997;275:1280–1304, with permission.) |
Myelomeningocele afflicts females slightly more commonly than males (57,70) and is evident at birth in all cases. The lesion involves the lower back predominantly: thoracic 2%, thoracolumbar 32%, lumbar 22%, and lumbosacral 44% (57). Because studies of embryos indicate that cervical and holocord myeloceles and myelomeningoceles actually occur more commonly than lumbosacral lesions, the predominance of caudal lesions evident at birth most likely reflects their lesser severity and greater survival to term (71).
Patients with myelomeningocele manifest a number of neurologic signs, including sensorimotor deficits of the lower extremities, incontinence of bladder and bowel, hindbrain dysfunction, intellectual–perceptual impairments, and hydrocephalus (72,73,74,75,76). A component of the motor deficit appears to result from birth trauma and spinal shock. Infants delivered by cesarean section before onset of labor manifest less severe deficits than those delivered vaginally or those delivered by cesarean section after some period of labor (77). Motor strength typically improves one to two levels after the initial repair of the myelomeningocele and then stabilizes at a set motor level. Deterioration thereafter is regarded as evidence of a secondary complication.
Recent decades have brought significant improvement in the care and prognosis of patients with myelomeningocele (78). In an older cohort born in 1975–1979, mortality at the 20- to 25-year follow-up was 24% and continued to increase into young adulthood. A majority of the deaths occurred during infancy and the preschool years. Most of the children died of hindbrain dysfunction. Shunt diversion of CSF was required in 86%; 95% of those with shunts required at least one revision (78).
In a younger cohort born in 2000–2004, none of the children died during infancy or in the preschool years (78). Four children underwent posterior fossa decompression. One child required a tracheostomy with long-term ventilator support (since weaned successfully), and four children required gastrostomy tubes (three removed when they later became unnecessary) (78). Only 65% required shunt diversion of CSF. Seventy-one percent have required one shunt revision (78).
Myelocele and myelomeningocele appear to result from deranged neurulation, although other theories have been offered to explain limited numbers of cases (38,79,80). If the neural folds fail to flex and to fuse into a tube, they persist instead as a flat plate of neural tissue (Fig. 19.14) (81,82). This flat plate of unneurulated neural tissue is designated the neural placode. Because the neural tube does not close, the superficial ectoderm cannot disjoin from the neural ectoderm and remains in lateral position. Therefore, the skin that develops from the ectoderm also lies lateral in position, leaving a midline defect. Mesenchyme then cannot migrate behind the neural tube, and so the bony, cartilaginous, muscular, and ligamentous elements are also deficient in the midline. Instead, the bones, cartilage, muscle, and ligament develop in an abnormal position ventral-lateral to the neural tissue and appear bifid and “everted.” The unfused neural plate is thus exposed to view in the midline of the back at the site of the midline deficiency of skin, bone, cartilage, muscle, and ligament.
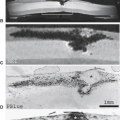
The exposed neural tissue appears as a raw, reddish, vascular oval plate (Fig. 19.15A). The raw surface represents the interior of what should have been the closed spinal cord. A midline
groove runs down the center of this plate. This groove is the residuum of the ventral neural groove and is directly continuous with the central canal of the normally formed cord above.
groove runs down the center of this plate. This groove is the residuum of the ventral neural groove and is directly continuous with the central canal of the normally formed cord above.
 FIGURE 19.14 Unneurulated neural placode. Proposed embryogenesis for spina bifida aperta by failure to neurulate the neural plate with consequent nondisjunction of epidermal from neural ectoderm. A: Mammalian embryo. B: Diagrammatic representation. Images oriented like axial magnetic resonance imaging (MRI) with ventral toward the top. (From Naidich TP, Gorey MT, Raybaud C, et al. Malformations congénitales de la moelle. In: Manelfe C, ed. Imagerie du rachis et de la moelle. Paris: Editions Vigot; 1989, with permission.) C: Resulting spina bifida with widely everted laminae. Axial T1 MRI in a 4-year-old girl. If the neural folds fail to close, the neural plate appears to open widely and to evert with the lateral margins ventral to the rest of the plate. The neural placode thus remains in a nearly coronal plane. The epidermal ectoderm (c) (large curved arrows) cannot disjoin from the neural ectoderm (NE). Instead, it remains attached to the neural ectoderm far laterally and does not pass dorsal to the neural tissue. With future development, the skin that arises from the epidermal ectoderm will also lie far laterally, leaving a midline defect in the skin cover. Adherence of the epidermal ectoderm to the neural ectoderm prevents migration of mesenchyme behind the neural plate (placode). Therefore, no bone, muscle, or fascia can develop in the midline dorsal to the neural tissue; all skin, fascia, muscle, and bone must lie lateral or ventral to the neural tissue. Thus, the dorsal surface of the unneurulated neural placode will remain visible in the midline. Despite absence of neurulation, the notochord on the ventral aspect will still migrate ventrally to become separated from the neural plate. Perichordal mesenchyme will develop into vertebral centra and disks ventral to the neural tissue. The mesenchyme that should have migrated dorsal to the neural tube must instead condense in abnormal position, ventral–lateral to the neural tissue, forming widely everted (bifid) laminae (L, L). C, vertebral centrum. Compare the contours of the bifid laminae with the mesenchyme ventral–lateral to the recurved neural placode. Because the laminae (L) are widely everted, the sagittal dimension of the spinal canal is markedly reduced, crowding the neural structures. Acutely angled kyphosis, often associated with wide spina bifida, stretches the skin and subcutaneous tissue, and prevents the surgeon from mobilizing enough tissue to cover the spina bifida. There is consequently little tissue and no high-signal subcutaneous fat overlying the spina bifida (arrowheads). The neurocentral synchondroses (white arrows) have nearly fused. (From Naidich TP, McLone DG. Congenital pathology of the spine and spinal cord. In: Taveras JM, Ferucci JT, eds. Radiology—Diagnosis/Imaging/Intervention. Philadelphia, PA: Lippincott; 1986, with permission.) |
The pia-arachnoid membrane that covers the ventral surface of the neural plate and encloses the cerebrospinal fluid (CSF) may bulge posteriorly to present at the surface of the back as a thin ring surrounding the neural tissue. The size of this membranous ring varies. When the subarachnoid space is small, the membranous ring is narrow, and the neural plate lies flush with the back. This situation is designated myelocele (Figs. 19.15 and 19.16A). When the subarachnoid space is very large, the membranous ring is wide, and the neural plate is elevated far above the skin surface. This condition is designated myelomeningocele (Figs. 19.15B and 19.16B). Together, myelocele and myelomeningocele are the most common forms of spinal dysraphism. When used colloquially, spina bifida usually means myelocele or myelomeningocele.
In both myelocele and myelomeningocele, the membranous ring itself is encircled by normal skin peripheral to the midline defect. Epithelial cells may later grow inward from the skin margins to cover the membranes and even the neural tissue that lie within the defect (Fig. 19.15B). If infection does not cause early death, the entire site may become epithelialized secondarily by a thin dysplastic skin layer.
Deep to the visible surface, the ventral face of the neural plate represents the neural tissue that should have formed the entire outer circumference of the spinal cord. The two ventral motor nerve roots arise from the ventral surface of the neural plate just to each side of the midline ventral sulcus. The paired dorsal sensory roots also arise from the ventral surface of the
neural plate, lateral to the corresponding ventral roots. They arise in this position because the tissue that should have folded dorsally to close the tube unfolds instead to lie lateral to the motor tissue on each side. The ventral and dorsal nerve roots traverse the subarachnoid space and exit via the neural foramina in the usual manner.
neural plate, lateral to the corresponding ventral roots. They arise in this position because the tissue that should have folded dorsally to close the tube unfolds instead to lie lateral to the motor tissue on each side. The ventral and dorsal nerve roots traverse the subarachnoid space and exit via the neural foramina in the usual manner.
 FIGURE 19.15 Myelocele and myelomeningocele. Photographs of the backs of two newborn patients. A: In myelocele, the neural placode lies nearly flush with the surrounding skin surface. The edges of the skin (white arrows) are separated from the edges of the neural plate (white arrowheads) by variably epithelized membranes. Note the midline groove and the two paramedian grooves (open white arrows). These would have formed the ventral neural groove and the sulci limitans of the ventral canal of a closed, well-neurulated spinal cord. B: In myelomeningocele, expansion of the subjacent subarachnoid space is associated with elevation of the neural plate well above the surrounding back. In this patient, skin partially encloses the meningocele, so the edge (black arrows) of the skin lies along the lateral margins of the neural plate with little intervening nonepithelialized membrane. Note the drop of cerebrospinal fluid (open arrow) that has passed from the central canal over the external surface of the neural placode and runs down the slope of the myelomeningocele. |
The pia-arachnoid membrane covers the ventral surface of the neural plate and continues around the entire subarachnoid space as one continuous sheet (Fig. 19.16) (81). This membrane is given the name pia mater where it is contiguous with neural tissue and the name arachnoid mater where it is separated from the neural tissue. The relationship of pia to arachnoid mater is analogous to the relationship of visceral to parietal pleura. The dura mater lies peripheral to the arachnoid. The dura forms a distinct layer ventrally but becomes lost in the margins of the defect dorsally. Because the neural plate and meninges
are anchored to the skin surface, the spinal cord is tethered and relatively immobile.
are anchored to the skin surface, the spinal cord is tethered and relatively immobile.
 FIGURE 19.16 Diagrammatic representation of myelocele and myelomeningocele. A: Myelocele. The myelocele is nearly flush with the surface of the back. The neural tissue has the flat configuration of the unneurulated neural placode (NP). The dorsal surface, exposed to the exterior, exhibits midline and paramedian sulci (arrows) corresponding to the ventral neural groove and sulci limitans (unless secondary trauma or infection distorts the tissue further). The ventral surface is lined by pia that becomes continuous with arachnoid at the lateral edge of the neural plate. This pia-arachnoid (dashed line) is one continuous sheet of membrane that encloses the subarachnoid space (SAS). The dorsal roots (D) arise from the ventral surface of the neural plate lateral to the ventral roots (V), not dorsal to them, because the plate has not flexed into a tube. G, the dorsal root ganglion. The pia-arachnoid continues along the ventral and dorsal roots but is not shown, to simplify the diagram. The dura (dark line superficial to the pia-arachnoid) encloses the cerebrospinal fluid ventrally and laterally and then becomes lost in the tissue lateral to the neural placode. No dura forms dorsal to the neural placode. The laminae (L) are widely everted (wide posterior spina bifida). B: Myelomeningocele. The myelomeningocele exhibits the same basic anatomy, but expansion of the SAS displaces the neural placode posteriorly, everts it, and elevates it well above the surface of the surrounding back. (From Naidich TP, Gorey MT, Raybaud C, et al. Malformations congénitales de la moelle. In: Manelfe C, ed. Imagerie du rachis et de la moelle. Paris: Editions Vigot; 1989; and Naidich TP, Gorey MT, McLone D. Congenital anomalies of the spine and spinal cord. In: Putman CE, Ravin CE, eds. Textbook of Diagnostic Imaging. Philadelphia, PA: WB Saunders; 1987, with permission.) |
 FIGURE 19.17 Extensive untreated myelomeningocele. Newborn. Sagittal T1-weighted (A) and T2-weighted (B) magnetic resonance imaging (MRI) and axial T1-weighted MRI (C). There is lumbosacral spina bifida with widely everted laminae and midline discontinuity of the subcutaneous fat, deep lumbosacral fascia, and posterior spinal elements (between white arrows). The low-lying distal cord (c, larger arrowheads) extends posteriorly beneath the last intact spinal element, opens into the neural plate, and becomes the thin posterior wall of the large myelomeningocele sac. Because the cord lies in low position, there is no cauda equina. Instead, the ventral and dorsal nerve roots (smaller arrowheads) arise from the ventral (deep) surface of the neural plate and pass nearly horizontally across the sac to reenter the spinal canal before exiting at their normal neural foramina. |
Rarely, one is able to study the untreated myelomeningocele (Figs. 19.17 and 19.18). Nearly always, however, patients with myelocele and myelomeningocele do not require detailed radiologic evaluation in the newborn period because the pathology is already evident on visual inspection. The back is simply repaired surgically to protect against infection and to free the spinal cord, so that later growth of the child does not cause neurologic dysfunction by cord stretching. As a result of this primary repair, the child’s neurologic function is stabilized or is improved to some “best” level for the child. Thereafter, the child should maintain that level of function (72).
Any subsequent deterioration in neurologic function represents a complication that must be evaluated for potential surgical correction. The usual role of radiology is to elucidate the cause for late deterioration in patients who have already undergone primary repair of the myelomeningocele. Such complications include retethering of the cord to the wall of the spinal canal, abnormal spinal curvature, inclusion epidermoid, inclusion lipoma, hydromyelia, and arachnoid cyst.
Devascularization of the Placode
In patients with myelomeningocele, the placode is supplied by radiculomedullary branches and by many large vessels that pass directly to the neural tissue via the lateral dural reflections. Surgery may injure these lateral dural vessels, especially superiorly at the junction between the neurulated cord and placode, leading to focal cord infarction and atrophy (1,30,73).
Local Wound Site
The most common complications at the site of the closure of the myelomeningocele include skin necrosis, CSF leak at the repair site (6% to 17%), CSF pseudocyst (2%), and local wound
infection (3% to 12%, higher for patients undergoing delayed closure) (57,74,83,84,85,86,87,88,89,90,91,92).
infection (3% to 12%, higher for patients undergoing delayed closure) (57,74,83,84,85,86,87,88,89,90,91,92).
 FIGURE 19.18 Untreated myelomeningocele. A: Patient photograph. The surface of the myelomeningocele has become covered by skin through secondary epithelialization from the margins of the defect. B: Sagittal T1-weighted magnetic resonance demonstrates features characteristic of untreated myelomeningocele: dehiscence of high-signal subcutaneous fat (F), fascia, muscle, and bone in the zone of spina bifida; low position of the spinal cord (C); acute angulation (arrow) of the cord under the last intact lamina at the upper margin of the spina bifida; and posterior herniation of the neural tissue (white arrowheads) forming the dorsal wall of the cerebrospinal fluid space (S) that protrudes through the spina bifida. (From Naidich TP, Radkowski MA, Britton J. Real-time sonographic display of caudal spinal anomalies. Neuroradiology 1986;28:512–527, with permission.) This patient also exhibits thoracolumbar kyphos. |
Latex Anaphylaxis
From 18% to 51% of patients with myelomeningocele (and up to 6% of medical personnel) show latex sensitivity, including latex antibodies in their sera (87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107). This occasionally leads to life-threatening anaphylaxis during surgery (87). Of 162 patients with latex allergy in Holzman’s study (90), 26% had previously manifested an anaphylactic reaction in the operating room. The powder on surgical gloves may be a vehicle for transmitting protein allergens from the latex gloves to the patients. Careful washing of gloves and careful histories of prior reactions need to be obtained to avoid these problems.
Retethering by Scar
When the spinal cord is freed from the back at the time of primary repair, it is replaced into the spinal canal. The dura and skin are closed over it. Because dura and skin are deficient in the midline, it may be technically difficult to mobilize sufficient tissue to provide a relaxed cover over the spinal canal, subarachnoid space, and cord. The raw surfaces of the neural tissue may adhere to the surgical closure and scar densely to it (Figs. 19.19 and 19.20) (91).
In approximately 27% of patients with repaired myelomeningocele, retethering of the cord by scar causes new symptoms that necessitate a second procedure to release the cord at an average age of 6 years (92). These symptoms include new or progressive weakness of the lower extremities (55%), change of gait requiring added mechanical support (54%), new or progressive scoliosis (51%), pain localized to the back and legs (32%), progressive orthopedic deformities of the foot or hip (11%), and/or new urinary incontinence (6%) (83).
In such cases, surgical photographs (Fig. 19.20) and radiologic studies (Figs. 19.21 and 19.22) show that the posterior surface of the spinal cord becomes lost at the level of closure (93). The spinal cord remains attached to the dorsal wall of the spinal canal despite placing the patient prone and flexing the back. The subarachnoid space is obliterated dorsal to the cord and is unusually wide ventral to the cord. The entire thecal sac lies unusually far posteriorly and protrudes partially through the posterior spina bifida. The ventral epidural fat space is consequently enlarged. The cord itself appears thin and pursues a very straight, stretched, “bowstring tight” course down the
length of the spinal canal. Because the cord extends so far caudal, there is no cauda equina. Instead, the nerve roots that arise from the cord at the level of the spina bifida pass horizontally or even cephalically to their root sleeves. These roots tend toward a plexiform arrangement (Fig. 19.20).
length of the spinal canal. Because the cord extends so far caudal, there is no cauda equina. Instead, the nerve roots that arise from the cord at the level of the spina bifida pass horizontally or even cephalically to their root sleeves. These roots tend toward a plexiform arrangement (Fig. 19.20).
 FIGURE 19.19 Repaired myelocele. Retethering by scar. Axial anatomic specimen oriented like magnetic resonance. The site of repair exhibits thinner atrophic skin with a thin subcutaneous layer (white arrows). The neural tissue (NP) is tethered to the surgical repair site by thick collagenous scar (black arrows). L, L, everted, widely bifid laminae. (From Naidich TP, Gorey MT, Raybaud C, et al. Malformations congénitales de la moelle. In: Manelfe C, ed. Imagerie du rachis et de la moelle. Paris: Editions Vigot; 1989, with permission.) |
 FIGURE 19.20 Repaired myelocele. Retethering by scar, 2-year-old girl. Sagittal surgical exposure demonstrates the low-lying distal spinal cord (C), horizontal segmental origin, and course of the lumbosacral roots through the subarachnoid space (S) with no cauda equina, somewhat plexiform arrangement of these roots, dorsal surface of the spinal canal along the zone of repair (R), adherence (black arrows) of the distal neural tissue to the repair zone at two sites, and clear cerebrospinal fluid–filled space behind the neural tissue between the two sites of adherence. |
Re-operation for tethering after primary myelomeningocele repair may be complicated by coexisting pathology. In 123 patients, Reigel et al. (94) found concurrent lipoma (50%), fibrolipoma (9%), dermoid (12%), epidermoid (16%), arachnoid cyst (11%), and diastematomyelia (11%). Approximately 10% of these patients had two concurrent lesions (94).
Surgical release of the adherent cord is effective, leading to improved motor function in 79% (including unexpected improvement in many patients who had been considered “stable” before untethering). Re-operated patients show improved gait and stance in 72% of cases, improvement of orthopedic deformities in 54%, improved scoliosis in 51%, and improved urinary and social continence in 33% (83). However, 16% of patients initially untethered later require surgical procedures to again untether a re-scarred cord (94).
Inclusion Cysts
(Epi)dermal inclusion cysts are found in both the unoperated newborn and the previously repaired back (74,97,98,99). Before closure of the myelomeningocele, the placode and central canal are open to the environment and exposed to desquamated skin, laguno, and hair in the amniotic fluid. Thus, Storrs (97) found epidermoid cysts in 29% and dermoid cysts in 6% of newborns during the initial closure of their myelomeningocele, including epidermoid “pearls” floating freely in the spinal subarachnoid space. McLone and Naidich (74) reported delayed occurrence of dermoid and epidermoid tumors in up to 16% of patients after myelomeningocele repair.
During the repair of a myelomeningocele, the surgeon must trim all the skin away from the cord, lest some skin elements become included within the closure. This is hard, probably impossible, to do perfectly. In some cases, therefore, these included (epi)dermal elements may later grow into epidermoid (12% to 13%) and dermoid cysts (2% to 5%) (Fig. 19.23) (85,97). Scott et al. (100) found that 15% of repaired myelomeningocele patients who later presented with a symptomatic tethered cord had a dermoid or epidermoid. In their experience, (epi)dermoids found within the scar, dorsal to the placode, were more likely to be inclusion (epi)dermoids resulting from surgery, whereas those found ventral to the neural tissue and those containing unusual features such as respiratory epithelium were more likely to be congenital in origin (100).
Lipomas are found in 6% of newborn untreated myelomeningoceles and 7% of operations performed to release tethered cords. Emery and Lendon (101) found filar fibrolipomas in 67% of myelomeningocele patients coming to postmortem. It may be difficult to distinguish inclusion lipomas from inclusion dermoids by imaging procedures alone (1,102,103,104).
Hydromyelia
Before the back is repaired, the central canal of the spinal cord is directly continuous with the midline groove on the dorsal surface of the neural plate. As a result, CSF passes freely down the central canal to discharge over the dorsal (external) surface of the
neural plate (Fig. 19.15B). After repair of the back, this egress for CSF is blocked, potentially leading to dilation of the central canal of the spinal cord (i.e., hydromyelia) (Fig. 19.24). Reviews of CSF pulsations (105,106) and of spinal cord cysts (107,108), however, suggest that hydromyelia may also be expected whenever the subarachnoid space is narrowed around the spinal cord and whenever the spinal cord is tethered, offering other possible mechanisms by which hydromyelia could arise.
neural plate (Fig. 19.15B). After repair of the back, this egress for CSF is blocked, potentially leading to dilation of the central canal of the spinal cord (i.e., hydromyelia) (Fig. 19.24). Reviews of CSF pulsations (105,106) and of spinal cord cysts (107,108), however, suggest that hydromyelia may also be expected whenever the subarachnoid space is narrowed around the spinal cord and whenever the spinal cord is tethered, offering other possible mechanisms by which hydromyelia could arise.
 FIGURE 19.21 A 15-year-old boy with uncomplicated prior repair of a sacral myelomeningocele. A: Midsagittal T2 MRI. B: Axial sacral T2 MRI. The spinal cord extends inferiorly through the lumbar canal and becomes adherent to the posterior wall of the canal along the surgically repaired sacral myelomeningocele (white arrow). |
 FIGURE 19.22 A 6-year-old boy with prior repair of a thoracic myelocele, atrophic segment of cord and hydromyelia. A: Midsagittal T2 MRI. B–D: Serial axial T2 MRIs. A: Following repair, the spinal cord demonstrates hydromyelia from T8–T10, an atrophic segment adherent to the posterior wall of the spinal canal at T10–T11, and a more nearly normal segment distally. |
 FIGURE 19.23 Repaired myelomeningocele with epidermoid inclusion in a 4-year-old child. Sagittal T1-weighted (A) and T2-weighted (B) magnetic resonance imaging disclose a repaired lumbosacral myelomeningocele. The low-lying spinal cord (c) is tethered inferiorly by a mass (arrowhead) that is nearly isointense to the cord on the T1- and T2-weighted images. Closure of the sac has created dorsal fluid compartments with restricted communication. Such compartments appear brighter on T2-weighted images because of reduced dispersal of signal by cerebrospinal fluid pulsations and perhaps because of higher protein concentrations within them. |
 FIGURE 19.24 Repaired myelomeningocele with hydromyelia. A 9-month-old boy. A: Sagittal T2 MRI. B: Axial T2 MRI. Cephalic to the repair (white arrows), the spinal cord is distended by hydromyelia (H). Jack-knife kyphus is also noted. |
Hydromyelia is observed at necropsy in 30% to 50% of patients with myelomeningocele (101,109,110,111,112). The precise incidence varies from series to series (101,112). The hydromyelia is holocord or nearly holocord in 11%, cervical in 2%, cervicothoracic in 7%, thoracic in 7%, thoracolumbar in 11%, and lumbar in 9%. Seven percent of patients show hydromyelia at two different levels (112).
When severe, hydromyelia may cause neurologic dysfunction and rapidly progressive scoliosis (113). In any patient with myelomeningocele and rapidly progressive scoliosis, cranial imaging studies should be performed to rule out hydrocephalus or occult shunt malfunction. Spinal imaging should be performed to detect any hydromyelia present. On occasion, a severe hydromyelia may expand the cord remarkably, thin its wall, and bulge outward partially exophytically, perhaps at its upper or lower pole (114). The hydromyelic cavity may appear to spiral around a portion of the circumference of the cord.
On MR, hydromyelia usually appears as a CSF-intensity space within the center of a dilated cord. The sides of the cavity may be indented periodically by bands that resemble the haustra of the colon. Hydromyelia may also present as an ovoid, sagittally flattened, atrophic cord (collapsed hydromyelia). In some cases, the central canal is so wide and the cord so thin that only one wall of the cord is visible, usually the anterior wall. In these cases the thin anterior wall may be mistaken for the entirety of a very atrophic spinal cord. In other cases it may be very difficult to distinguish hydromyelia with scoliosis from intraspinal arachnoid cyst. Such arachnoid cysts are found in 2% of patients with myelomeningocele (Fig. 19.25) (26,115).
Diastematomyelia and Hemimyelocele
Diastematomyelia is seen in 31% to 46% of patients with myelomeningocele (76,79). It affects the cord cephalic to the plaque in 31%, the plaque itself in 22%, and the cord caudal to the plaque in 25% (101). Hemimyelocele is a special form of myelomeningocele-with-diastematomyelia that is reported in 9% of patients with Chiari II malformations (116). In 1%, paired hemicords each exhibit a myelomeningocele at different levels. In the other 8%, one posteriorly situated hemicord exhibits a myelomeningocele, whereas the other hemicord is normal. The normal hemicord is usually smaller, usually lies ventrally, and may be isolated within a separate ventrolateral hemicanal formed by an oblique bone spur. In such cases, the visible hemimyelocele may be mistaken for a complete myelomeningocele, so that the diastematomyelia and other hemicord are not recognized until the child presents with tethering sometime after the initial repair. Strikingly asymmetric motor strength in the lower extremities, with one normal or nearly normal leg, should suggest the diagnosis of hemimyelocele in the newborn period.
Spinal Curvature in Myelomeningocele
The incidence of spinal curvature in patients with myelomeningocele depends on the level of the myelomeningocele, patient age, and the type of curvature assessed (Fig. 19.16, Table 19.1) (117,118,119,120,121).
TABLE 19.1 Spinal Curvature and Myelomeningocele | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
 FIGURE 19.25 Cervicothoracic arachnoid cyst with Chiari II malformation. A: Sagittal T1-weighted magnetic resonance imaging (MRI) reveals the Chiari II malformation with kink and spur where medulla (M) buckles behind the cord (C) (see Figs. 19.28 and 19.29). The tapering of the cord inferiorly could indicate scoliosis alone, hydromyelia with thin undetectable posterior wall, and/or dorsal arachnoid cyst. B: Axial T1-weighted MRI demonstrates a large low-signal space posterior to the cord (C) and suggests that the space does not represent hydromyelia at that level. Cord contour is slightly “dumbbell” shaped. C: Computed tomographic myelography reveals a less well-communicating compartment dorsal to the cord representing a subdural arachnoid cyst (A). The cord exhibits a mild form of diastematomyelia. D: Operative exposure in another patient, a 3-year-old boy, with Chiari II malformation and myelomeningocele. The dura is tented laterally by stay sutures. The thin-walled, transparent, sausage-shaped arachnoid cyst lies within the subdural space and displaces the spinal cord anteriorly. It contained clear fluid resembling cerebrospinal fluid. E: Opening the cyst reveals the spinal cord (C) situated ventral to the cyst. The dorsal surface of the spinal cord was deeply indented by the cyst. (Panels D and E: From Naidich TP, McLone DG, Fulling KH. The Chiari II malformation. Part IV. The hindbrain deformity. Neuroradiology 1983;25:179–197, with permission.) |
Scoliosis
In patients with myelomeningocele, 23% of the scolioses are congenital structural scolioses that result from vertebral anomalies such as hemivertebrae, solid bony bars, and unilateral unsegmented bars. In such cases, the arc of curvature is very sharp and the apex of the curve lies at the site of the bony anomaly (1,122). Sixty-five percent of scolioses are developmental scolioses that result from muscle imbalance and distal pelvic and extremity malformations in the absence of congenital spinal anomalies (122). In these patients the arc of curvature is usually less sharp and lies in the dorsolumbar region.
The incidence of scoliosis associated with myelomeningocele varies with the functional spinal level of the patient: 85% for thoracic myelomeningocele, 100% for L1–L2 myelomeningocele, 50% for L3–L4 myelomeningocele, and 6% for sacral myelomeningocele (123). Overall, 80% of patients with myelomeningocele have scoliosis by age 10 years (92). Progression of scoliosis occurs in the first decade and dramatically increases during the second decade for patients with functional levels at the thoracic or upper lumbar (L1–L3) spine (94). After 10 years of age, 43% of myelomeningocele patients have scoliosis of 20 degrees or greater (87).
Although spinal curvatures are widely thought to be most rapidly progressive at the time of growth spurts, careful study of spinal curvatures in 23 patients receiving growth hormone showed no significant change in the scoliotic curve and no evidence of neurologic change over 3 years, despite patient growth at a rate of 7 to 8 cm/year (94).
In patients with myelomeningocele, retethering of the spinal cord should always be considered the likely cause of scoliosis. In these patients, surgical untethering of the cord stabilizes most cases and actually improves the curve in 21% of patients (92). The greatest incidence and greatest severity of scoliosis occur with thoracic myelomeningoceles (94). These are also the most refractory to therapy (94).
Lumbar Lordosis
In normal patients, the usual lumbar lordosis develops rapidly during infancy and again at puberty, reaching a plateau of about 50 degrees at maturity. Significant lordosis, defined as more than 55 degrees, develops by age 20 years in 43% of patients with repaired myelomeningocele and tethered cord. As with scoliosis, more-cephalic lesions show a greater incidence of lordosis and a more rapid progression. After release of the tether, the improvement of lordosis is greatest for those patients with severe thoracic myelomeningocele.
Thoracic Kyphosis
In myelomeningocele patients, a thoracic kyphosis develops slowly and “normally” reaches 40 degrees by 20 years. Thoracic kyphosis of more than 40 degrees is seen in 21% of patients with tethered cord after myelomeningocele repair and exhibits the same craniocaudal gradient of incidence and severity as do scoliosis and lumbar lordosis. However, release of the cord does not seem to affect the progression and incidence of thoracic kyphosis.
Lumbar Kyphosis
Congenital lumbar kyphosis is a major deformity in 8% to 21% of patients with myelomeningocele and is frequently associated with compensatory thoracic lordosis (118,124,125). It is usually located in the upper lumbar region, measures 80 degrees or more at birth, and progresses thereafter. The vertebra at the apex of the curve is typically hypoplastic and anteriorly wedged (Fig. 19.26) (1). The aorta lies well away from the kyphus and in 56% is strung like a bowstring across the kyphotic vertebral curve (126). The lumbar arteries originating from the aorta follow a long horizontal course from the aorta to the dorsum of the kyphos and are commonly variant (126,127). In one case, for example, a single trunk arose from the aorta distal to the renal arteries and ascended close to the vertebrae, giving rise to two intercostal arteries per segment over five lumbar segments.
In patients with lumbar kyphosis, the sharp curvature leads to tight stretching of the skin over the closure, skin ulceration, breakdown, and infection. These patients suffer progressive respiratory difficulty because of incompetence of inspiratory muscles, crowding of abdominal content, and upward pressure on the diaphragm (124). Excision of the proximal lordosis stabilizes their curvature and facilitates ambulation. Children born with a thoracolumbar kyphos who function at or below L4 should be considered for kyphectomy before their second birthday (92).
Carstens et al. (125) distinguished three subtypes of lumbar kyphosis in myelomeningocele: paralytic (less than 90 degrees at birth), sharp angled (greater than 90 degrees at birth), and congenital (resulting from structural malformations of the vertebrae, such as anterior vertebral malsegmentation). Paralytic kyphosis has nearly normal lumbar spinal curvature at birth but cannot maintain this because of muscle weakness. Such paralytic kyphosis accounts for 44% of lumbar kyphosis and progresses linearly with later development. Sharp-angled kyphosis typically has a more rigid curvature caused by the pathologic position of the paraspinal muscles. It accounts for 38.4% of lumbar kyphosis and also progresses linearly with growth. In both types, the actual progression also depends on the level of paralysis. Congenital kyphosis accounts for 13.9% of lumbar kyphosis and shows variable progression (125).
Effect of Untethering on Spinal Curvature
Sarwark et al. (128) studied the effect of surgery on the spinal curvature of 29 myelomeningocele patients operated on to release retethering. At 1 year after untethering, 75% of patients
had stabilized their curvature or had improved it by greater than 10 degrees; 25% showed more than 10-degree progression of their curvature (128). Continued follow-up revealed that only 12% maintained an improvement in curvature of greater than 10 degrees; 47% remained stable, and 41% progressed at least 10 degrees (128). Reigel et al. (94) similarly assessed the effect of surgical untethering on spinal curvature in 216 myelomeningocele patients. Progression of scoliosis plateaued or declined after release of the tethered cord for patients with myelomeningoceles at the lumbar and sacral levels, but cord release did not halt the progression of scoliosis in patients with thoracic level lesions. Untethering improved the course of lordosis in patients with lesions at the L1–L3 levels but not in patients with L4, L5, or sacral-level lesions. Untethering did reduce the incidence and magnitude of kyphosis (94).
had stabilized their curvature or had improved it by greater than 10 degrees; 25% showed more than 10-degree progression of their curvature (128). Continued follow-up revealed that only 12% maintained an improvement in curvature of greater than 10 degrees; 47% remained stable, and 41% progressed at least 10 degrees (128). Reigel et al. (94) similarly assessed the effect of surgical untethering on spinal curvature in 216 myelomeningocele patients. Progression of scoliosis plateaued or declined after release of the tethered cord for patients with myelomeningoceles at the lumbar and sacral levels, but cord release did not halt the progression of scoliosis in patients with thoracic level lesions. Untethering improved the course of lordosis in patients with lesions at the L1–L3 levels but not in patients with L4, L5, or sacral-level lesions. Untethering did reduce the incidence and magnitude of kyphosis (94).
 FIGURE 19.26 A 2-month-old infant with repaired myelomeningocele below T7 (see also Fig. 19.30A). A: Reformatted sagittal CT scan. There is “jack-knife” kyphus of the lumbar spine. The subcutaneous fat, fascia, muscle, and bone are markedly thinned in the zone of spina bifida. B: Corresponding 3D reformation of the spine. C: Midsagittal T2 MRI. The spinal cord (arrows) thins out into the zone of repair. |
TABLE 19.2 Indications, Incidence and Outcomes for Untethering a Retethered Cord in 114 Patients with Repaired Myelomeningocele and Cord Retethering | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
Cord Retethering and Release of the Retethered Cord
Bowman et al. (78) analyzed the incidence of spinal cord retethering and the effect of detethering the cord in a large, single-institution cohort of myelomeningocele patients. Symptomatic retethering of the spinal cord was found in 114 of 502 children (23%) with myelomeningocele who underwent initial closure at that institution. The 114 children underwent 163 procedures for release of tethered cord: 81 children (71%) had one procedure, 20 (17%) had two procedures, 10 (9%) had three untetherings, and four (3%) had four. Indications and outcomes for untethering the retethered cord are given in Table 19.2. Caldarelli et al. (129) and Hayashi et al. (130) report similar findings in mixed populations of patients with repaired myelomeningoceles and repaired spinal lipomas.
Fetal Repair of Myelomeningocele
Increasingly, myelomeningoceles are being detected prenatally during fetal maternal sonography and then confirmed by subsequent fetal–maternal MRI (Fig. 19.27). This raises consideration of fetal surgery to repair the defect in utero. The Management of Myelomeningocele (MOM) study compared the results of prenatal surgery for myelomeningocele repair at 19 to 25 weeks of gestation (WG) versus postnatal surgery by the same team following delivery at 37 WG (131). Overall, the children in the group operated upon prenatally presented with higher, more severe lesions: 27% of lesions at L1 or L2 in the prenatal group versus 12% in the postnatally operated group (131). Nonetheless, this study found that prenatal surgery reduced the incidence of fetal and neonatal death and reduced the need for shunt therapy of hydrocephalus. The prenatally treated group had reduced incidence of hindbrain herniation, reduced severity of any hindbrain herniation present,
motor function that was one to two levels better than predicted from lesion level of myelomeningocele, better Bayley Mental Development Index, and similar Wee FIM cognitive scores (a measure of pediatric functional independence) (131). Twice as many children treated prenatally were walking independently, and fewer were not walking at all (131). The study was terminated prematurely for early documentation of benefit from the prenatal surgery.
motor function that was one to two levels better than predicted from lesion level of myelomeningocele, better Bayley Mental Development Index, and similar Wee FIM cognitive scores (a measure of pediatric functional independence) (131). Twice as many children treated prenatally were walking independently, and fewer were not walking at all (131). The study was terminated prematurely for early documentation of benefit from the prenatal surgery.
 FIGURE 19.27 Fetal–maternal T2 MRIs. Two patients. A: Lumbar myelomeningocele (red arrow) at 25 weeks gestation. B: Thoracic myelomeningocele (red arrow) at 27 weeks gestation. |
In a second study of 54 patients who underwent fetal repair of myelomeningocele, 16 (30%) presented with symptomatic tethered cord syndrome 4 to 93 months (median 27 months) after repair (132). Ten of the 16 (19% of the total) had an intradural inclusion cyst as part of the tethered cord syndrome (132). Four additional children (7% of the total) had evidence of an inclusion cyst on MRI but were asymptomatic at 60 to 94 months following repair (132). Histologically, eight resected cysts were dermoids, one an epidermoid, and one a mixed dermoid–epidermoid cyst (132).
Fetal surgery does carry risk of adverse effects. Because of the uterine surgery, the mothers of the prenatal group are more likely to develop pulmonary edema, placental abruption, oligohydramnios, spontaneous rupture of membranes, or spontaneous labor (131). At the time of cesarean delivery, 25% of the women who underwent fetal surgery had a very thin hysterotomy site. Nine percent had an area of dehiscence along the scar, and 1% had complete dehiscence of the incision (131). To reduce these risks, the children must be delivered prematurely, with all the attendant risks of prematurity. Ultimately 8% of the children in this group required surgery for tethered cord versus 1% in the group operated upon postnatally (131).
Chiari II Malformation
Myelocele and myelomeningocele are nearly always associated with a specific deformity of the brainstem, cerebellum, and upper cervical spinal cord, designated the Chiari II malformation (Fig. 19.28) (26,115,133,134,135). This malformation presents clinically with hindbrain dysfunction. Historically, the hindbrain dysfunction was severe in 4% to 13% of all patients with myelomeningocele (136) (7% of infants and 2% of older patients) (75,136). Recent decades have brought significant improvement in the prognosis of patients with myelomeningocele (78). In an older cohort born in 1975 to 1979, mortality at the 20- to 25-year follow-up was 24% and continued to increase into young adulthood. A majority of the deaths occurred from hindbrain dysfunction during infancy and the preschool years (78). In a younger cohort born in 2000–2004, none of the children died during infancy or in the preschool years, and few required posterior fossa decompression, tracheostomy, or gastrostomy (78).
The specific clinical signs of hindbrain dysfunction show very similar frequencies in different series (Table 19.3). Apnea may result from bilateral abductor vocal cord paralysis (obstructive apnea), from central neural dysfunction (centrally mediated expiratory apnea with cyanosis) (138), or from both together.
Multiple theories have tried to explain the diffuse manifestations of the Chiari II malformation and myelomeningocele
(26,139). McLone and Knepper (139) presented experimental data suggesting that the Chiari II malformation could be the result of diversion of ventricular CSF to the amnion with consequent “collapse” of the developing ventricular system. The McLone–Knepper theory is based on the following facts:
(26,139). McLone and Knepper (139) presented experimental data suggesting that the Chiari II malformation could be the result of diversion of ventricular CSF to the amnion with consequent “collapse” of the developing ventricular system. The McLone–Knepper theory is based on the following facts:
The fluid-filled space of the developing brain and spinal cord is called the neurocele.

FIGURE 19.28 Chiari II malformation. Medullary protrusion and the cervicomedullary kink in an 11-month-old boy. A: Left lateral view of the uncut hindbrain. B: Midsagittal section of the same brain (larger field of view includes corpus callosum). Anterior is to the reader’s left. White arrows indicate the position of foramen magnum. Large white arrowhead indicates the site of the posterior arch of C1. The Chiari II medulla (M) protrudes below foramen magnum and C1 into the cervical spinal canal. The medulla buckles dorsal to the cervical spinal cord and forms a kink (crossed white arrow), where it forces the upper cervical cord to recurve on itself and forms a spur (double-crossed white arrow) at the true cervicomedullary junction. The medullary hernia overlaps the cervical cord. The fourth ventricle (small white arrowheads) is greatly elongated, extends into the cervical canal, and widens there. Choroid plexus (open white arrowheads) lies along the dorsal aspect of the intraspinal fourth ventricle. The vermis protrudes downward through the large foramen magnum, rests on the posterior arch of C1 (large white arrowhead), and extends a tongue or peg of tissue (V) through the C1 ring dorsal to the medulla and fourth ventricle. Note the sharp notch (large white arrowhead) where the posterior lip of the C1 ring indents the vermian peg. The cerebellar hemispheres (H) pass anteriorly to lie in front of and encompass the brainstem. Also demonstrated are the beaked midbrain (m) and the pons (P). (From Naidich TP, McLone DG, Fulling KH. The Chiari II malformation. Part IV. The hindbrain deformity. Neuroradiology 1983;25:179–197, with permission.)
TABLE 19.3 Clinical Signs of Chiari II Hindbrain Dysfunction
Sign
Park et al. (113) (n = 45a)
Vandertop et al. (137) (n = 17a)
Pollack et al. (267) (n = 13a)
Swallowing difficulty
69%
71%
92%
Stridor/vocal cord dysfunction
56%
59%
69%
Apneic spells
58%
29%
54%
Aspiration
40%
12%
—
Weak cry
—
18%
—
Arm weakness
27%
53%
15%
Opisthotonus
18%
—
46%
aAll operated on.
The medial walls of the thoracic neural tube normally appose and occlude the neurocele transiently during central nervous system (CNS) development in humans, mice, and chicks.
Experimental animals with distal myelomeningocele fail to occlude the neurocele, even at sites remote from the myelomeningocele.
This failure to appose the walls appears to result from the same biosynthetic defect in cell-surface glycosaminoglycans which prevents the neural tube from closing. That is, the mechanism that causes failure of neurulation also causes failure of apposition of the medial walls of the neurocele. Based on these findings, the theory proceeds as follows.
Because the neurocele is not occluded, CSF passes freely down the central canal and out the myelomeningocele to the amnionic cavity.
This abnormal shunt collapses the developing primitive ventricular system.
Therefore, the volume of the ventricular system and surrounding neural tissue is less than normal.
The mesenchyme condenses in relation to an abnormally small volume of developing CNS. This establishes a smaller-than-normal posterior fossa with low tentorium.
The developing CNS must then grow within an envelope of membrane, cartilage, and bone that is too small for it.
This leads to failure to form the pontine flexure, downward growth of the cervicomedullary junction, medulla, and cerebellum through the foramen magnum, and upward growth of the cerebellum through the incisura.
Reduced size of the third ventricle means closer approximation of the thalami with larger massa intermedia.
Collapse of the cerebral ventricles leads to disorganization of the developing hemispheres with gray matter heterotopias, disorganization of cerebral gyri, and dysgenesis of the corpus callosum.
The collapse of the ventricular system leads to disordered development of the membranous bone of the vault (139). Normally, the skull develops from centers in each cranial plate. As the brain expands, the collagen bundles are drawn out from those centers in an orderly radial fashion, much like the uniform expansion of the surface of an inflating balloon. As radial expansion proceeds, the collagen bundles become calcifiable and membranous bone forms. Failure to distend the brain mass by increasing volumes of CSF causes disordered arrays of collagen bundles. Thus, instead of radial lines of collagen, whorls, and coils of collagen form with varying density between them. Ossification of this disorganized collagen mat then leads to lükenshädel.
In accord with the McLone–Knepper hypothesis, a study of 56 children with repaired myelomeningocele documented that the smaller size of the Chiari II posterior fossa and the reduced volume of the Chiari II cerebellum are both directly and linearly related to the level of the myelomeningocele (140). Further, the extent to which the cerebellum descends into the spinal canal is inversely related to the level of the myelomeningocele. These relationships strengthen in patients with no syringomyelia and are not significant in those groups with syringomyelia (140).
In the Chiari II malformation the upper cervical cord is displaced caudally, so that the cervical nerve roots ascend retrograde to their exit foramina. The brainstem is also displaced caudally, so that the medulla and even the low pons lie within the cervical spinal canal.
The medulla may descend purely vertically, so it remains in line above the spinal cord (30%). More commonly, the medulla buckles backward and downward as it descends, forcing the dorsal surface of the uppermost cervical cord to kink back on itself (the “kink”) and forming a spur of tissue at the cervicomedullary junction (the “spur”) (70%) (Figs. 19.28 and 19.29). The cuneate and gracile tubercles at the upper ends of the dorsal columns form the apex of the spur and point caudally, not cephalically.
Emery and MacKenzie (115) demonstrated that the deformities at the cervicomedullary junction form a spectrum of pathology that falls into five major groups. In order of increasing severity (Fig. 19.29), these can be categorized as follows:
Group A: In 4% of Chiari II patients the medulla and fourth ventricle do not descend below foramen magnum. The sole evidence of hindbrain deformity is mild inferior displacement of the spinal cord with ascending course of the cervical nerve roots.
Group B: In 26% of cases, the medulla and fourth ventricle descend vertically, in line with and above the displaced cervical cord. In these cases the fourth ventricle leads directly inferiorly into the central canal of the cord (Fig. 19.30).
Group C: In another 26% of cases the medulla shows mild buckling behind the cervical cord, so there is less than 5 mm of overlap of medulla on cord. Because of the buckling, the fourth ventricle lies partly behind the cord, and the central canal arises from the anterior surface of the cord.
 FIGURE 19.29 Diagrammatic representation of the spectrum of cervicomedullary deformities in the Chiari II malformation. Lateral views oriented with anterior to the reader’s left. A–E: Graded series of increasingly more severe deformities discussed fully in the text. Curved lines at the top indicate the foramen magnum. Black shading indicates the fourth ventricle and central canal of the spinal cord. (From Emery JL, MacKenzie N. Medullo-cervical dislocation deformity Chiari II deformity related to neurospinal dysraphism meningomyelocele. Brain 1973;96:155–162, with permission.) |
Group D: In 23% of cases the medulla shows greater buckling, with greater than 5 mm overlap of medulla behind the cord (Figs. 19.25A and 19.28A).
Group E: In a further 21% of cases, severe buckling of medulla is associated with a saclike process or diverticulum of the dorsal surface of the fourth ventricle. This protrudes behind the cord caudal to the kink (Fig. 19.31).
The fourth ventricle descends with the brainstem and forms a craniocaudally elongated tubular structure that lies partially within the posterior fossa and partially within the spinal canal. The choroid plexus of the fourth ventricle may protrude inferior to the cerebellar tail to present as a contrast-enhancing nodule (141). Structural lesions may be found within the Chiari II fourth ventricle, including arachnoid cysts, glial or choroidal nodules, and subependymomas (142). These lesions may be developmental or may arise in reaction to the chronic compression and ischemia (142).
 FIGURE 19.30 Chiari II malformation in a 9-month-old boy with myelomeningocele (see also Fig. 19.29B). Sagittal T2 MRI of the low posterior fossa and upper cervical spinal canal displays prominent concavity of the basiocciput (BO), low position of the medulla (M), thin elongated fourth ventricle (4), and protrusion of the vermis into the spinal canal below C3 (3). BS, basisphenoid; P, pons. |
 FIGURE 19.31 Chiari II malformation in a 2-month-old one with myelomeningocele (see also Fig. 19.29A). Sagittal T2 MRI displays a dilated third ventricle (3), kinked aqueduct with beaked tectum (black arrow), thin elongated fourth ventricle (4), caudal protrusion of the brainstem and vermis into the spinal canal, and a cystic elongation (c) of the 4th ventricle into the spinal canal, inferior to the vermis. Note inferior wall of cyst (white arrow), separate from the subarachnoid space. M, midbrain; P, pons. |
The cerebellum usually protrudes caudally into the spinal canal behind the medulla. The major portion of the cerebellum that protrudes below the foramen magnum cannot fit into the C1 ring and appears to rest or “sit” on the posterior arch of C1. A smaller, thinner portion of cerebellum, designated the cerebellar “peg,” “tongue,” or “tail,” does extend further caudally within the C1 ring. Typically, the peg is formed by the nodulus (most inferiorly), the uvula (more superiorly), and the pyramis (often situated within the foramen magnum). The length of the peg is variable and does not correlate with the length of the medullary spur. In 75% of cases, the cerebellar tail terminates cephalic to the kink (Fig. 19.28). In 25%, the tail extends below the kink, behind the lower cervical cord.
The overlapping herniations of cerebellum on medulla and of medulla on cord form a “cascade” of hernias, each of which displaces anteriorly and compresses all structures ventral to it. All the herniations are compressed in turn by impaction in foramen magnum and the small C1 ring. Because the tentorium is low in position, the midbrain is subjected to pressure from the distended hydrocephalic atria. This bilateral lateral pressure molds the tectum of midbrain into a conical “beak.” Numerous other anatomic distortions, also present, are delineated elsewhere (26,134,135,143,144).
Curnes et al. (144) reported that 75% of myelomeningocele patients with symptomatic Chiari II malformation manifested a spur at C4 or below. No asymptomatic patient exhibited a spur lower than the C3–C4 level (144). Gilbert et al. (143) found
evidence of brainstem dysplasia in 76% of 25 symptomatic myelomeningocele children evaluated pathologically at postmortem, including foci of abnormal myelination and hypoplasia/aplasia of cranial nerve nuclei, basal pontine nuclei, the olivary nuclei, and the tegmentum. The anatomic changes of the Chiari malformations are summarized in Cesmebasi (145).
evidence of brainstem dysplasia in 76% of 25 symptomatic myelomeningocele children evaluated pathologically at postmortem, including foci of abnormal myelination and hypoplasia/aplasia of cranial nerve nuclei, basal pontine nuclei, the olivary nuclei, and the tegmentum. The anatomic changes of the Chiari malformations are summarized in Cesmebasi (145).
Cervical Myelomeningocele and Myelocystocele
The cervical myelomeningocele and myelocystocele are two lesions that appear to arise by a common mechanism. Many authors (16,45,146,147,148,149,150) have suggested that these malformations develop when there is incomplete fusion of the neural folds, causing a narrow dorsal myeloschisis and incomplete disjunction of epidermal from neural ectoderm (Fig. 19.32). As a result, there is a weak, incomplete dorsal neural wall that is still adherent to the epidermal ectoderm. If there is significant hydromyelia, dilation of the central canal might cause the thinned dorsal wall of the cord to balloon backward. Because the meninges form in relation to the external surface of the neural tube, they would surround the ballooned neural tube, except at the dorsal midline. This would create a meningeal sac around the posteriorly bulging neural tube. The mesenchyme would then condense and mature around these structures, leaving a narrow spina bifida, through which the neural tissue would appear to herniate into the meningocele sac. A lesion of this type is designated cervical myelocystocele (syringocele).
If there is no hydromyelia, the dorsal neural–epidermal connection might involute to a fibroglioneural stalk. Nearly complete involution of the stalk could lead to a form of meningocele manqué. Variably complete involution of the stalk and outward bulging of the meninges might form a meningocele that is traversed by a fibroglioneural stalk. Condensation and development of the mesenchyme around such a complex could then result in a skin-covered cervical myelomeningocele. With variation, the stalk could insert into the apex, the base, or one wall of the meningocele, and fibroglioneural nodules could persist along the wall of the sac.
The pathogenesis of these cervical malformations closely resembles the pathogenesis of dorsal dermal sinuses (discussed in the next section). In cervical myelomeningoceles and myelocystoceles, a partial neural–epidermal adhesion draws the neural tissue outward toward the skin, whereas in dermal sinuses, a partial neural–epidermal adhesion draws the skin inward toward the neural tissue. Both types of lesions may exhibit partial dorsal myeloschisis. To date, however, no unifying theory has been elaborated to explain all of these malformations coherently.
Children with cervical myelocystocele form a special group of myelomeningocele patients (3.7%) who present with a tough, skin-covered midline cervical mass and (nearly) normal neurologic function (151). The base of their lesion is covered by full-thickness skin, whereas the dome is covered by thick squamous epithelium (151). The spinal cord remains within the spinal canal but is tethered to the myelomeningocele by a midline or paramedian fibrous septum or by a band composed of neurons, glia, fibrous tissue, and peripheral nerves (Fig. 19.33) (146). The fibroglioneural band passes through a narrow cervical spina bifida to insert into small glial islands or into glial–neural disks along the adjacent dura or intrasaccular soft tissue. Treatment of these lesions requires opening the dura, with lysis of all adhesions and fibroneurovascular connections, lest the children deteriorate in the years after surgery (146).
Clinically, these two groups of patients present differently. Patients with cervical myelomeningocele show no obvious neurologic deficits at birth, although they may develop mild orthopedic problems (147). Patients with myelocystocele develop motor deficits in the first year and show significant orthopedic problems, although sensory and urologic deficits are rare (147). The myelocystocele may be associated with Chiari II malformation (70), hydrocephalus, ectopic cerebellar tissue, and diastematomyelia (151,152).
Dorsal Dermal Sinus
If the superficial ectoderm fails to separate from the neural ectoderm at one point, then a focal segmental adhesion is created. As the spinal cord becomes buried beneath the surface by the developing spinal column and as different rates of growth between neural and spinal tissue lead to “ascent” of the cord, the local adhesion or “spot-weld” is drawn out into an elongated epithelial-lined tube that still connects the spinal cord with the skin of the dorsal surface of the child (Fig. 19.34) (1,5,153). Such an elongated segmental epithelial tract is designated a dorsal dermal sinus. The points of attachment of skin and cord remain segmental or metameric, and so the dermatome of involvement predicts the site of neural abnormality. In Wright’s collected series of 127 dermal sinuses (154), 57% were lumbosacral and 24% were occipital. The rest occur at uncommon sites.
Dermal sinuses are found in 1 per 1,500 to 2,500 newborns (155,156,157). Males and females are affected equally. Clinically, the sinus most frequently appears as a pinpoint hole or a small atrophic zone in the skin. The sinus ostium is typically midline but may be paramedian in unusual cases. A tuft of short, sparse, wiry hairs may emerge from the ostium (153). Small hemangiomas commonly adjoin or surround the ostium (Fig. 19.35).
Typically, the dermal sinus tract extends inward from the skin surface for a variable depth (Fig. 19.36). The deep end of the dermal sinus/stalk most commonly ends at the conus (9/14, 64%) or intradural portion of the filum terminale (4/14, 29%), but may end “short” at the dura mater (1/14, 7%) (158). In its course the dermal sinus passes deeply through the subcutaneous layer and through the median raphe (or between bifid laminae) toward the dura. It may end superficial to the dura, at the dura, or deep to the dura. In one-half to two-thirds of cases, the tract extends into the spinal canal (1,154,155,159). A small midline sleeve of dura and arachnoid, directed dorsally, marks the point at which the tract attaches to or penetrates the dura (Fig. 19.36). Rarely, the tract ends in the subarachnoid space as an open tube through which CSF discharges and through which infection may ascend retrograde to the spinal canal (159). Such infection may lead to arachnoiditis, epidural/subdural abscess, and intramedullary spinal cord abscess (160,161,162). The dermal sinus may also end in a fibrous nodule among the roots of cauda equina. In up to 60% of cases, the tract incorporates or ends in one or multiple dermoid or epidermoid tumors (Fig. 19.37). Approximately 25% of all spinal (epi)dermoids are associated with dermal sinuses (16). The (epi)dermoids may act as masses. Cyst rupture may cause sterile chemical meningitis and obliterative arachnoiditis.
Lumbosacral dermal sinuses are usually associated with tethering of the spinal cord and low position of the conus (80%) (16,24). Thoracic and cervical dermal sinuses do not appear to influence conal position but may still produce symptoms by local cord tethering (163). Some 15% to 20% of patients with dermal sinus have concurrent lipoma, and vice versa, presumably because both lesions result from deranged disjunction of cutaneous from neural ectoderm (Fig. 19.38). Such an error in disjunction could lead to failed disjunction (dermal sinus), premature disjunction (spinal lipoma), or both at adjacent sites.
Imaging studies typically demonstrate the small depression in the skin at the ostium. The extraspinal portion of the dermal sinus tract may appear as a single line or as double parallel (railroad) lines that traverse the midline and correspond to the lumen and walls of the tract (164). Infrequently, the tract is filled by the included (epi)dermoid. Lumbosacral sinuses
typically first course inferiorly and ventrally as they pass from the skin surface to the lumbodorsal fascia. They then abruptly reverse direction to course superiorly as they pass further ventrally into the spinal canal (Figs. 19.38 and 19.39). Less commonly, the superficial portion of the tract appears horizontal or even ascends from the skin toward the spine. The precise course may depend on the degree of lumbar lordosis and the position of each sinus with respect to that curvature.
typically first course inferiorly and ventrally as they pass from the skin surface to the lumbodorsal fascia. They then abruptly reverse direction to course superiorly as they pass further ventrally into the spinal canal (Figs. 19.38 and 19.39). Less commonly, the superficial portion of the tract appears horizontal or even ascends from the skin toward the spine. The precise course may depend on the degree of lumbar lordosis and the position of each sinus with respect to that curvature.
 FIGURE 19.32 Cervical myelomeningocele and cervical myelocystocele: proposed embryogenesis. Diagrammatic representations. The two forms of cystic cervical malformations begin as limited dorsal myeloschisis of the developing cervical cord and evolve differently in response to the presence or absence of hydromyelia. A: Limited dorsal myeloschisis. The dorsal neural tube fuses incompletely, and the epidermal ectoderm disjoins incompletely from the neural ectoderm. B,C: Cervical myelocystocele. Hydromyelia (B) distends the central canal and bulges it posteriorly. Subarachnoid cerebrospinal fluid forms a meningocele around the entire neural tube, except where the partly closed neural tube is adherent to the epidermal ectoderm in the midline. This results in a cervical myelocystocele in continuity with the central canal of the cord (C), within a meningocele that is continuous with the subarachnoid space. Condensation and development of the perineural mesenchyme create a narrow spina bifida traversed by the thinned, ballooned, dorsal wall of the neural tube and the surrounding meninges. D,E: Cervical myelomeningocele. In the absence of hydromyelia, the dorsal wall of the partly closed neural tube begins to regress and narrow, leaving a dorsal neural stalk that is still adherent to the surface (D). The subarachnoid space bulges dorsally to produce the meningocele. This results in an intracanalicular spinal cord tethered to the wall of the meningocele (E), either in the midline as shown here or at the base of the meningocele sac. Compare with the embryogenesis and development of the dorsal dermal sinus (Figs. 19.34–19.37). (Modified from Sun JCL, Steinbok P, Cochrane DD. Cervical myelocystoceles and meningoceles: long term follow-up. Pediatr Neurosurg 2000;33:118–122; Steinbok P, Cochrane DD. Cervical meningoceles and myelocystoceles: a unifying hypothesis. Pediatr Neurosurg 1995;23:317–322; and Steinbok P. Dysraphic lesions of the cervical spinal cord. Neurosurg Clin N Am 1995;6:367–376, with permission.) |
 FIGURE 19.33 Cervical meningocele in an untreated 1-day-old boy. A: Sagittal T1-weighted magnetic resonance imaging (MRI). B: Axial T1-weighted MRI. No Chiari II malformation is evident. The low cervical meningocele traverses a narrow midline spina bifida (arrowhead). The adjacent portion of the cervical cord is enlarged sagittally and pointed posteriorly, indicating tethering to the sac. Inhomogeneous signal within the sac represents pulsations and proteinaceous material; no neural tissue was found within the sac at surgery. |
The intraspinal portion of the tract is hard to identify. It may be indistinguishable from the filum or from the roots of cauda equina unless it is expanded by (epi)dermoids or contains fat (16).
In one series of 28 dermal sinuses, females predominated (F:M = 17:11) (165). The sinuses were most commonly found in the lumbosacral (9), lumbar (9), thoracic (5), and cervical regions (4). One dermal sinus at L3 was associated with an additional coccygeal pit at L5. In one patient, two cutaneous openings overlay a single tract (165). Cutaneous stigmata were present in 27 of the 28 cases (165). Nineteen of the 28 cases (68%) had neurological deficit on initial examination, including motor weakness (11), sensory change (7), reflex changes (15), gait changes (5), decreased sphincter tone (6), or difficulty with bladder and bowel function (4) (165). Infection was present in three: two with infected skin lesions and one with a history of H. influenza meningitis (Fig. 19.39) (165). Patients with dermal sinuses commonly show concurrent vertebral anomalies (Table 19.4) (165).
 FIGURE 19.34 Proposed embryogenesis of dorsal dermal sinus by incomplete disjunction. Diagrammatic representation. Focal failure of epidermal ectoderm (c) to disjoin from neural ectoderm (NE) results in a persistent segmental adhesion (arrows). As the mesenchyme migrates dorsally to thicken the retroneural tissue and as the cord ascends, this adhesion is drawn out into a long epithelial-lined tube. M, mesenchyme; cc, central canal. (From Naidich TP, Gorey MT, Raybaud C, et al. Malformations congénitales de la moelle. In: Manelfe C, ed. Imagerie du rachis et de la moelle. Paris: Editions Vigot; 1989, with permission.) |
Recently, there has been an impetus to distinguish “true” dermal sinus tracts from dermal sinus–like stalks (166). True dermal sinus tracts are held to consist of a hollow, epithelium-lined tract lined by stratified squamous epithelium. This extends inward from the skin surface for a variable distance. Three key features
of true dermal sinuses are (1) an open skin defect, (2) the common concurrence of (epi)dermoid inclusions along the tract, and (3) frequent infections (166). Conversely, dermal sinus–like stalks exhibit a skin dimple or a translucent layer of thinned skin, with no ostium (166,167). They have no lumen and no concurrent inclusion cysts. They consist instead of mesodermal elements such as connective tissue, fat, muscle, and nervous–dural elements. Because there is no hollow lumen, infection is very rare (166). Further, in two large series, the conus medullaris was found to be in low position more commonly with a dermal sinus–like stalk (90.5%) than a true dermal sinus (38.5%) (158,167). The embryogenesis of such stalks is not yet determined.
of true dermal sinuses are (1) an open skin defect, (2) the common concurrence of (epi)dermoid inclusions along the tract, and (3) frequent infections (166). Conversely, dermal sinus–like stalks exhibit a skin dimple or a translucent layer of thinned skin, with no ostium (166,167). They have no lumen and no concurrent inclusion cysts. They consist instead of mesodermal elements such as connective tissue, fat, muscle, and nervous–dural elements. Because there is no hollow lumen, infection is very rare (166). Further, in two large series, the conus medullaris was found to be in low position more commonly with a dermal sinus–like stalk (90.5%) than a true dermal sinus (38.5%) (158,167). The embryogenesis of such stalks is not yet determined.
 FIGURE 19.35 Dorsal dermal sinus in a 1-month-old boy. Posterior view of the patient reveals the typical midline lumbosacral hemangioma. The hemangioma surrounds a small atrophic dimple that was the external end of the dermal sinus in this patient. These stigmata lie well above the anus and coccyx. |
 FIGURE 19.36 Dorsal dermal sinus. Diagrammatic representation. Note the cutaneous ostium, variable uncommon presence of a small tuft of wiry hairs protruding from the ostium, the course of the tract to the lumbodorsal fascia through the median raphe (or between bifid spinous processes), and passage of the tract through the dura and arachnoid, leaving a small triangular dorsal outpouching of the meninges akin to a root sleeve. The tract may then ascend within the spinal canal, incorporate one or several (epi)dermoid lesions, and end (variably) in an (epi)dermoid at the conus. The ascent of the extraspinal component of the sinus tract shown here is unusual in imaging studies. It may represent, in part, Matson’s experience with placing the patient in a prone flexed position for surgery. In our experience, imaging studies routinely show that the extraspinal component of the tract passes inferiorly and ventrally to the lumbodorsal fascia, before the deeper portion turns upward to ascend within the spinal canal (see Fig. 19.34). (From Matson DD. Neurosurgery of infancy and childhood. 2nd ed. Springfield, IL: Charles C Thomas; 1969, with permission.) |
 FIGURE 19.37 Dorsal dermal sinus in a 22-month-old boy. Pathologic specimen resected in toto. From the skin surface (arrowheads), the tract passed ventrally through the subcutaneous fat to the lumbodorsal fascia, traversed the spina bifida, pierced the dura (arrow), ascended within the thecal sac, expanded into a dermoid (curved arrow) within the distal thecal sac, and then continued as a tract toward the conus, where it was ligated and resected. |
 FIGURE 19.38 Dorsal dermal sinus and lipoma in a young child. Sagittal T1 (A) and T2-weighted (B) magnetic resonance imaging (MRI). The superficial portion (arrowheads) of the dermal sinus descends from the skin surface to the deep lumbodorsal fascia (small arrowheads). The sinus then penetrates the fascia, traverses the sacral spina bifida, and inserts into the inferior aspect of the spinal cord, tethering it. A concurrent spinal lipoma (Li) lies along the dorsal aspect of the cord at L4–L5, just superior to the insertion of the dermal sinus. C: Axial T1-weighted MRI at L4–L5. The lipoma (Li) lies solely along the dorsal surface of the tethered cord. |
 FIGURE 19.39 Dorsal dermal sinus and infection in a young child. Sagittal T1-weighted (A,B) and T2-weighted (C) magnetic resonance images reveal the oblique descending course of the superficial portion of the dermal sinus (arrowhead), slightly low position of the conus medullaris, and obliteration of much of the distal thecal sac by inflammatory debris. Ascending inflammatory changes alter the texture and signal of the distal cord. |
Coccygeal Dimples
Coccygeal dimples (synonyms: coccygeal pits, sacral dimples, pilonidal sinuses) are midline or paired-paramedian depressions and pits found in the skin overlying the low sacrum or coccyx
in 2% to 6% of infants (Figs. 19.40 and 19.41) (155,168,169,170,171). They are classified as shallow if the bottom is visible, and deep if the bottom is not visible (172). Deep dimples typically extend inward for a variable distance and may reach to the dorsal surface of the coccyx (Fig. 19.42). In one series, shallow dimples and deep gluteal creases were seen in 4.3% of 1997 consecutive term newborns (155). Deep dimples were seen in an additional 1.4% of newborns (155). Analysis of 3,884 high-resolution (8 to 14 MHz) spinal sonograms performed for sacral dimple at two hospitals over a 12-year period disclosed 133 cases (3.4%) abnormal for low cord position, decreased motion of cord or roots, or abnormal filum terminale. Only 5 of these 3,844 patients (0.13%) were judged to merit surgical intervention, revealing four tethered cords and one enlarging ventriculus terminalis with reduced cord motion (173). Martinez-Lage et al. (167) found concurrent separate coccygeal pits in 3 of 8 children with true dermal sinus tracts, but no coccygeal pits in 12 children with dermal sinus–like stalks. De Vloo et al. (158,165) found a coccygeal pit in one of five patients (20%) with dermal sinuses and one of nine patients (11%) with dermal sinus–like stalks. Similarly, Ackerman and Menezes (165) found a coccygeal pit in 1 of 28 patients (4%) with dermal sinus. However, Harada’s (172) review of MR scans in 84 patients with sacral dimples revealed concurrent lipoma of the filum terminale in 14 patients (16.7%), 5 of 58 with shallow dimples (8.6%), and 9 of 26 with deep dimples (34.6%). For these reasons, we use spinal sonography to scan such patients during the first 6 weeks of life, when possible, or MR thereafter.
in 2% to 6% of infants (Figs. 19.40 and 19.41) (155,168,169,170,171). They are classified as shallow if the bottom is visible, and deep if the bottom is not visible (172). Deep dimples typically extend inward for a variable distance and may reach to the dorsal surface of the coccyx (Fig. 19.42). In one series, shallow dimples and deep gluteal creases were seen in 4.3% of 1997 consecutive term newborns (155). Deep dimples were seen in an additional 1.4% of newborns (155). Analysis of 3,884 high-resolution (8 to 14 MHz) spinal sonograms performed for sacral dimple at two hospitals over a 12-year period disclosed 133 cases (3.4%) abnormal for low cord position, decreased motion of cord or roots, or abnormal filum terminale. Only 5 of these 3,844 patients (0.13%) were judged to merit surgical intervention, revealing four tethered cords and one enlarging ventriculus terminalis with reduced cord motion (173). Martinez-Lage et al. (167) found concurrent separate coccygeal pits in 3 of 8 children with true dermal sinus tracts, but no coccygeal pits in 12 children with dermal sinus–like stalks. De Vloo et al. (158,165) found a coccygeal pit in one of five patients (20%) with dermal sinuses and one of nine patients (11%) with dermal sinus–like stalks. Similarly, Ackerman and Menezes (165) found a coccygeal pit in 1 of 28 patients (4%) with dermal sinus. However, Harada’s (172) review of MR scans in 84 patients with sacral dimples revealed concurrent lipoma of the filum terminale in 14 patients (16.7%), 5 of 58 with shallow dimples (8.6%), and 9 of 26 with deep dimples (34.6%). For these reasons, we use spinal sonography to scan such patients during the first 6 weeks of life, when possible, or MR thereafter.
TABLE 19.4 Radiographic and Operative Findings in 28 Patients with Dermal Sinus | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 FIGURE 19.40 Sacral dimple in a 1-month-old girl. An ostium situated near to the anus may be a “sacral” dimple or a dorsal dermal sinus. |
 FIGURE 19.41 Deep coccygeal dimple in a 3-year-old girl. The tract extends caudally to reach the coccyx. |
 FIGURE 19.42 Sacral dimple in a 2-month-old girl. Axial T1-weighted magnetic resonance imaging demonstrates that the dimple and tract (arrow) run directly to the coccyx (c) and thus that the lesion is a sacral dimple, not a dermal sinus tract. |
Unless infected, sacral dimples cause no difficulty and are not directly associated with cord tethering or intraspinal anomalies (171). If infected, sacral dimples may cause painful local abscesses, requiring drainage or excision. Uncommonly, spread of infection from such an abscess may lead to meningitis and even death (155).
Dermoid and Epidermoid Tumors
Traditionally, a distinction has been made between epidermoid and dermoid cysts. Epidermoid cysts are masses lined by a membrane composed only of superficial (epidermal) elements of the skin. Dermoid cysts are unilocular or multilocular cystic masses lined by a simple or stratified squamous epithelium that contains skin appendages such as hair follicles, sweat glands, and sebaceous glands. More recently, it has been held that both epidermoids and dermoids derive from ecto dermal elements (174). These (epi)dermoid tumors may arise as congenital rests in the absence of dermal sinuses (Fig. 19.43) or as the result of iatrogenic implantation of viable skin elements during back surgery or during spinal taps performed with needles without a trocar (1). Approximately 25% of (epi)dermoid tumors form in association with dermal sinuses (1,160).
Spinal dermoid and epidermoid tumors constitute about 15% of all CNS (epi)dermoids, a cranial-to-spinal ratio of 6:1 (103). They account for 1% to 2% of spinal cord tumors in patients younger than the age of 15 years (175). Dermoid and epidermoid tumors occur equally frequently (175). Approximately 40% are single epidermoids, 35% are single dermoids, and 5% are multiple dermoid or epidermoid tumors. In the series of Lunardi et al. (175), epidermoids were most frequently lumbar in location; dermoids were most frequently thoracic (25%) or thoracolumbar (75%). Thirty-eight percent were intramedullary, and 63% were intradural extramedullary (175).
Of (epi)dermoids associated with dermal sinus, approximately 60% have both intramedullary and intradural extramedullary components (16). In van Aalst’s (176) series of 18 (epi)dermoids, 5 were associated with dermal sinuses, 9 with repaired myelomeningoceles, 1 with spinal lipoma, 2 with unspecified dysraphisms, and 1 (5.6%) with no concurrent malformation. By location, 2 were thoracolumbar, 2 lumbar, 10 lumbosacral, and 4 sacral (176). One was intraspinal extradural, nine were intradural, and eight intramedullary. All those associated with dermal sinuses were lumbosacral (176).
Of (epi)dermoids associated with dermal sinus, approximately 60% have both intramedullary and intradural extramedullary components (16). In van Aalst’s (176) series of 18 (epi)dermoids, 5 were associated with dermal sinuses, 9 with repaired myelomeningoceles, 1 with spinal lipoma, 2 with unspecified dysraphisms, and 1 (5.6%) with no concurrent malformation. By location, 2 were thoracolumbar, 2 lumbar, 10 lumbosacral, and 4 sacral (176). One was intraspinal extradural, nine were intradural, and eight intramedullary. All those associated with dermal sinuses were lumbosacral (176).
 FIGURE 19.43 Intraspinal epidermoids in a 12-year-old boy. Sagittal magnetic resonance images: T1-weighted image (A), proton density-weighted image (B), and T2-weighted (C) image. Well-defined ovoid masses are related to the dorsal aspect of the conus medullaris at T12–L1 and to the cauda equina at L3–L4. No dermal sinus was identified on imaging studies or at surgery. D: Operative exposure after opening the dura in the midline reveals the two separate epidermoids (black arrows). |
Most dermoid and epidermoid tumors are homogeneously isointense to CSF or spinal cord on T1- and T2-weighted MRI. Specific analysis of signal intensity in 17 noninfected (epi)dermoids showed that on T1-weighted images 7 lesions
were isointense to CSF, 8 were isointense to spinal cord, and 2 were hyperintense to spinal cord. On T2-weighted images, 13 lesions were isointense to CSF and 4 isointense to spinal cord. Five of the 17 tumors were isointense to CSF on both T1- and T2-weighted images (176). A few (epi)dermoids showed heterogeneous signal (176). For these reasons, (epi)dermoids may be difficult to discern within the spinal canal. Intramedullary components do stand out in relation to the expanded cord (16,177), but extramedullary components may be invisible or poorly detectable on routine MR series, though readily demonstrable by diffusion-weighted sequences (16). Fat-containing portions of dermoids will typically manifest as lesions with high signal on T1 series and reduced signal on T2 series (178). Infected (epi)dermoids exhibit intense contrast enhancement within the tumor and at other sites to which the inflammation may spread (160,179,180).
were isointense to CSF, 8 were isointense to spinal cord, and 2 were hyperintense to spinal cord. On T2-weighted images, 13 lesions were isointense to CSF and 4 isointense to spinal cord. Five of the 17 tumors were isointense to CSF on both T1- and T2-weighted images (176). A few (epi)dermoids showed heterogeneous signal (176). For these reasons, (epi)dermoids may be difficult to discern within the spinal canal. Intramedullary components do stand out in relation to the expanded cord (16,177), but extramedullary components may be invisible or poorly detectable on routine MR series, though readily demonstrable by diffusion-weighted sequences (16). Fat-containing portions of dermoids will typically manifest as lesions with high signal on T1 series and reduced signal on T2 series (178). Infected (epi)dermoids exhibit intense contrast enhancement within the tumor and at other sites to which the inflammation may spread (160,179,180).
Spinal Lipoma
By definition, spinal lipomas are distinct collections of fat and connective tissue that are at least partially encapsulated and have a definite connection with the spinal cord (1,27,181,182). They exhibit a gradient of fat and fibrous tissue, with larger proportions of fat superficially and larger proportions of fibrous tissue deeply, closer to the liponeural junction. Spinal lipomas are the most common type of occult spinal dysraphism and account for 35% of skin-covered lumbosacral masses (Fig. 19.44). Typically, the mass lies in the midline just cephalic to the intergluteal crease (Fig. 19.44) and extends caudally, asymmetrically, into one buttock.
Spinal lipomas are found in approximately 1 per 4,000 newborns (183,184). Nearly all cases are sporadic. Familial spinal lipomas are exceptionally rare (183,185,186). The lipomas are equally common in males and females (187).
The chief complaints are usually a mass on the back (59%), urinary incontinence (23%), or weak/deformed lower extremity, perhaps with trophic ulceration (11%) (Table 19.5) (188). In combined series of 177 spinal lipomas, physical examination disclosed a soft tissue mass in 100%, skin dimple in 23%, hemangioma in 19%, hairy patch in 7%, skin tag in 6%, hypopigmented patch in 3%, and atretic/denuded skin patch in 0.6%
(189). Those patients with prominent subcutaneous lipomas and cutaneous stigmata (Table 19.6) are usually identified at a young age. Patients who are “missed” early on may present at older ages with neurologic deterioration (189,190).
(189). Those patients with prominent subcutaneous lipomas and cutaneous stigmata (Table 19.6) are usually identified at a young age. Patients who are “missed” early on may present at older ages with neurologic deterioration (189,190).
 FIGURE 19.44 Spinal lipoma in a 1-month-old girl. Photograph of the patient’s back reveals a skin-covered lumbosacral mass situated cephalic to the intergluteal cleft in the midline and extending asymmetrically to the reader’s right. (Same patient as in Fig. 19.51.) (From Naidich TP, McLone DG, Mutleur S. A new understanding of dorsal dysraphism with lipoma (lipomyeloschisis): radiological evaluation and surgical correction. AJNR Am J Neuroradiol 1983;4:103–116, with permission.)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|