• Congenital hepatic fibrosis is always present in patients with autosomal recessive polycystic kidney disease (ARPKD)
Sometimes with autosomal dominant PKD
Hepatic fibrosis that exists separately from other liver & renal disease is very rare
• Congenital hepatic fibrosis is variable in severity, age at presentation, and clinical manifestations
Portal hypertension is usually present by adolescence
• Diagnosis: Coexisting hepatic and renal cysts
Liver biopsy
• Treatment for moderate to severe fibrosis: Transplantation (hepatic and renal)
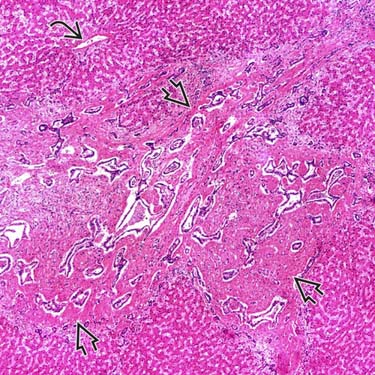
(Left) Low-power view shows marked portal expansion and numerous irregularly shaped bile ducts . The lobular architecture in adjacent parenchyma is well maintained with normal central veins . (Courtesy H. Wang, MD, PhD.)
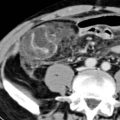
(Right) Axial CECT in a young woman with congenital fibropolycystic disease shows a dysmorphic, heterogeneous liver with several large cystic lesions, representing cystic and fusiform dilation of biliary tree (Caroli disease); note the central dot sign of Caroli disease.
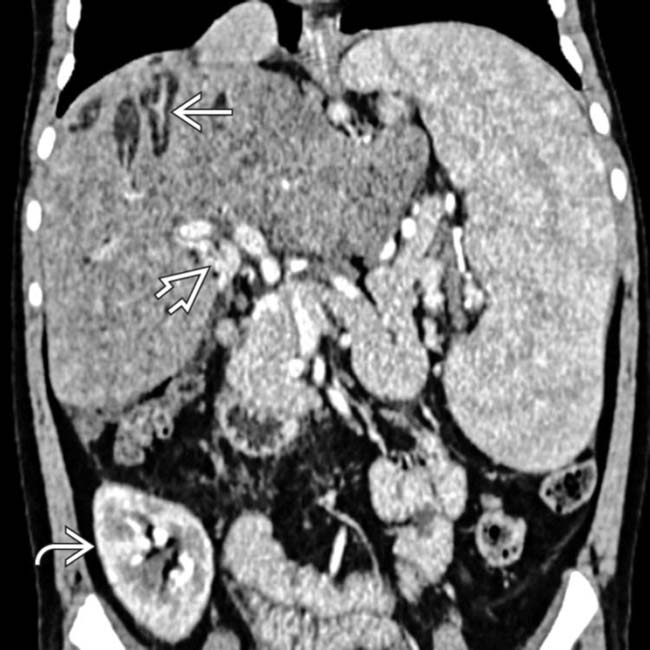
(Left) Axial CECT in a 40-year-old woman shows numerous cysts within the liver, many with the central dot sign that represents the portal vein branches around which the biliary cystic spaces of Caroli disease are wrapped. This patient had biopsy-proven congenital hepatic fibrosis, which results in hepatic failure and accounts for splenomegaly .
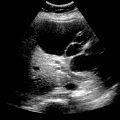
(Right) Coronal CECT in the same patient shows the cystic bile ducts of Caroli disease with large supernumerary hepatic arteries and a renal allograft .
TERMINOLOGY
Abbreviations
• Congenital hepatic fibrosis (CHF)
Definitions
• Part of a spectrum of congenital abnormalities resulting in variable degrees of fibrosis and cystic anomalies of liver and kidneys
• 1 manifestation of fibropolycystic liver disease
IMAGING
General Features
• Best diagnostic clue
Dysmorphic liver with cysts, abnormal ducts, + signs of portal hypertension
– May show similar changes within kidneys
• Location
Both lobes of liver
• Size
Hepatic cystic size varies based on severity of CHF
• Other general features
CHF is always present in patients with autosomal recessive polycystic kidney disease (ARPKD) and sometimes present with autosomal dominant PKD
2 constant features of ARPKD
– Kidney: Tubular ectasia, cysts, and fibrosis
– Liver: Fibrosis (dilated bile ducts, enlarged/fibrotic portal triads) and multiple cysts
All patients with ARPKD have findings of hepatic fibrosis on biopsy
Not all patients with hepatic fibrosis have ARPKD
Relative severity of disease may vary
– Renal or hepatobiliary disease may predominate
Variants of fibropolycystic liver disease
– CHF, biliary hamartomas, polycystic liver disease, choledochal cyst, and Caroli disease often coexist in same patient
Hepatic fibrosis that exists separately from other liver and renal diseases is very rare
Radiographic Findings
• ERCP
± dilatation of intrahepatic bile ducts
CT Findings
• Mild CHF
Liver may appear normal
• Moderate to severe fibrosis
Bile ducts: Normal to irregularly dilated
Enlarged dysmorphic liver, left lobe hypertrophy, right lobe atrophy
Splenomegaly and varices
Increased size and number of hepatic arteries
May develop hypervascular, benign, large, regenerative nodules (nodular regenerative hyperplasia)
– Similar to those seen in Budd-Chiari syndrome
• Associated polycystic disease of liver and kidney
Multiple hypodense (water density) hepatic and renal cysts of varied size
CECT: No enhancement of cyst contents
MR Findings
• MR cholangiography (MRC)
Bile ducts; variable pattern: Normal, irregular dilation, Caroli pattern
Only gold members can continue reading. Log In or Register to continue









 . The lobular architecture in adjacent parenchyma is well maintained with normal central veins
. The lobular architecture in adjacent parenchyma is well maintained with normal central veins  . (Courtesy H. Wang, MD, PhD.)
. (Courtesy H. Wang, MD, PhD.)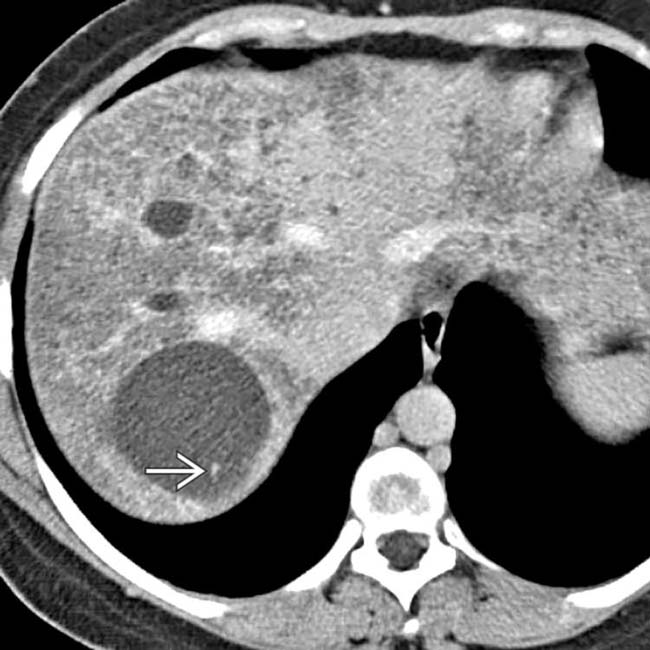
 of Caroli disease.
of Caroli disease.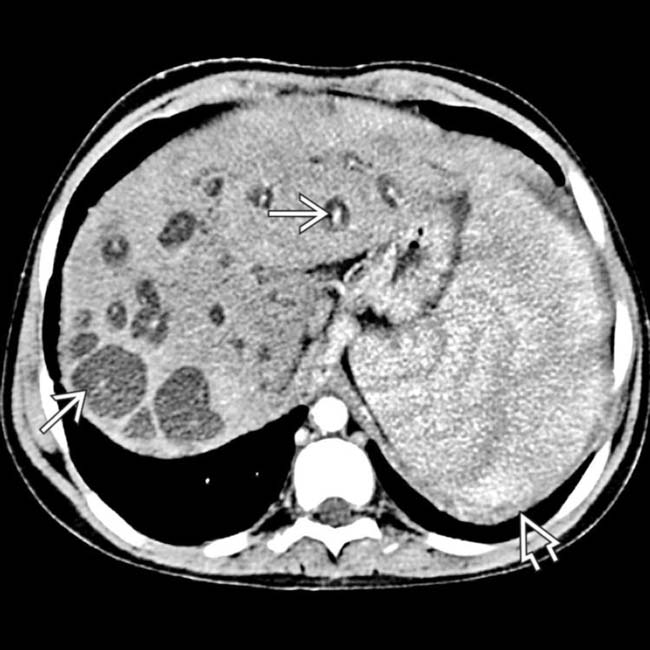
 that represents the portal vein branches around which the biliary cystic spaces of Caroli disease are wrapped. This patient had biopsy-proven congenital hepatic fibrosis, which results in hepatic failure and accounts for splenomegaly
that represents the portal vein branches around which the biliary cystic spaces of Caroli disease are wrapped. This patient had biopsy-proven congenital hepatic fibrosis, which results in hepatic failure and accounts for splenomegaly  .
.
 with large supernumerary hepatic arteries
with large supernumerary hepatic arteries  and a renal allograft
and a renal allograft  .
.