Fig. 1
A 14-year-old boy with cylindrical bronchiectasis in the middle lobe. Bronchi are dilated and slightly irregular
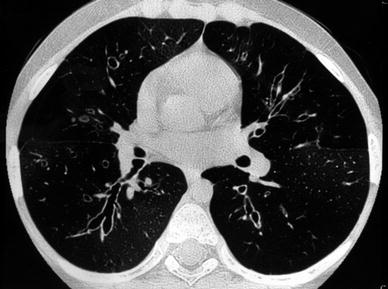
Fig. 2
An 11-year-old boy with varicose bronchiectasis. In the right lower lobe. Bronchi have an irregular and beaded appearance
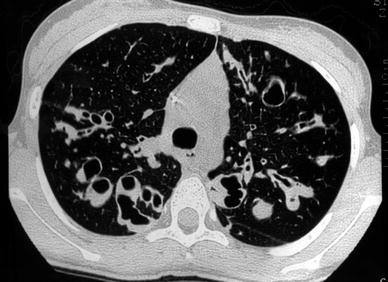
Fig. 3
A 14-year-old girl with cystic bronchiectasis. Saccular bronchiectasis with retained secretions are seen in both upper lobes
Retained bronchial secretions, atelectasis, and/or mosaic perfusion, can also be seen in HRCT scans of patients with bronchiectasis. When the bronchus is perpendicular to the scan plane, retained bronchial secretions in the central lung appear as nodular or oval-shaped opacities. When the bronchus is parallel to the scan plane, they are recognized as lobulated linear or branching structures. In the peripheral bronchi, retained mucus is visualized as centrilobular nodules or tree-in-bud appearance (Grenier et al. 1990). Mosaic perfusion, secondary to air-trapping and reflecting the presence of small airway disease distal to the bronchiectasis, is extremely common.
The pattern and distribution of abnormalities revealed by HRCT in patients with bronchiectasis are influenced by the underlying cause, and distinctive HRCT appearances have been well described in a few conditions: cystic fibrosis, immotile cilia, allergic asthma, bronchopulmonary aspergillosis, tuberculosis, and hypogammaglobulinemia (Cartier et al. 1999).
Bilateral, predominantly upper lobe bronchiectasis is seen most commonly in patients with cystic fibrosis and allergic bronchopulmonary aspergillosis, unilateral upper lobe predominance in patients with tuberculosis, and lower lobe predominance as a sequela of childhood pulmonary infections.
1.1.1 Cystic Fibrosis
Cystic fibrosis is the most common cause of pulmonary insufficiency in childhood (Ruzal-Shapiro 1998). HRCT and High-resolution MDCT technique have been used for the evaluation of cystic fibrosis lung disease. HRCT technique allows lower dose CT scanning and may be useful for qualitative evaluation. HRCT images at interval greater than 10 mm underestimate the severity of the disease (de Jong et al. 2006). High-resolution MDCT is recommended for longitudinal evaluation and for quantitative evaluation (Brody et al. 2005). This opinion is not accepted by all authors. They still prefer to reduce the dose using preselected CT cuts (Jiménez et al. 2006). Bronchiectasis in cystic fibrosis is usually widespread, with upper lobe involvement being almost universal and both central and peripheral bronchiectasis being present in approximately two-thirds of patients. Although cystic and varicose types are not uncommon, cylindrical bronchiectasis usually predominates, particularly in young children. Peribronchial thickening, mucoid impaction, and a mosaic perfusion pattern secondary to air-trapping are common in cystic fibrosis (Fig. 4). Mosaic perfusion may be the initial (and only) HRCT abnormality early in the course of the disease. Mucoid impaction can present as large nodules in the central lung or as centrilobular or tree-in-bud pattern in the lung periphery. Mucus plugging may lead to lobar and segmental atelectasis. Partial or total resolution of mucus plugging is a finding that can reflect therapeutic efficacy and is useful for monitoring.
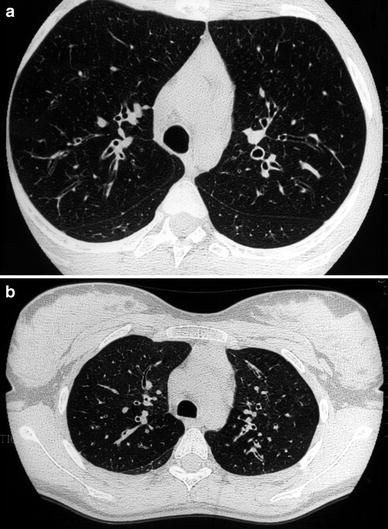
Fig. 4
A 15-year-old girl with cystic fibrosis. HRCT detects cylindrical bronchiectasis with bronchial wall thickening (a). Expiratory air-trapping reflects the presence of small airways disease (b)
Several authors have devised scoring systems based on chest X-ray findings to assess the severity of the disease (Nathanson et al. 1991; Sockrider et al. 1994; Cleveland et al. 1998). The Brasfield method is one of the most commonly used (Brasfield et al. 1980). New scoring systems based on HRCT have been proposed (Shah et al. 1997; Brody et al. 1999; Helbich et al. 1999). The most popular is the Bhalla method (Bhalla et al. 1991), which attempts to provide an objective assessment of the severity and extension of lung disease. Dissociation between CT score and lung function was reported by authors (de Jong et al. 2004; Brody et al. 2004). HRCT is a more sensitive technique than pulmonary function tests to detect structural changes and disease progression. However, the clinical relevance of HRCT scoring systems has not yet been demonstrated (Brody et al. 2005). Low-dose HRCT is now considered as an appropriate method for the evaluation of bronchiectasis in cystic fibrosis pediatric patients (O’Connor et al. 2010). But one must be aware that MRI gains increasing importance in the assessment and scoring of cystic fibrosis lung disease (Eichinger et al. 2012).
1.1.2 Immotile Cilia
Immotile cilia or primary ciliary dyskinesia (PCD) is a term including diseases that occur as a direct result of congenital defects in the airway cilia (Meeks and Bush 2000). The main features of PCD are recurrent sinopulmonary infections, situs inversus, and subfertility. The association between chronic respiratory disease and PCD is well-recognized. Kartagener’s syndrome, characterized by situs inversus totalis, bronchiectasis, and paranasal sinusitis accounts for 50 % of all patients with PCD (Hiddema and Engelshove 1999). The radiological and clinical features of PCD are similar to those of cystic fibrosis but are less severe and progressive. Hyperinflation and bronchial thickening are the most common abnormalities. Bronchiectasis (particularly in the right middle lobe), mucus plugging (Fig. 5), atelectasis, and consolidation are also frequent (Nadel et al. 1985; Fauré et al. 1986; Reyes de La Rocha et al. 1987). HRCT is currently used to monitor the disease progression despite the fact that CT monitoring affects poorly pulmonary outcome in PCD (Barbato et al. 2009).
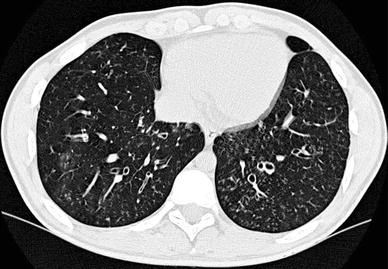
Fig. 5
A 15-year-old girl with primary ciliar dyskinesia. Cylindrical bronchiectasis with mucus plugging (tree-inbud pattern) is seen in both lower lobes
1.2 Asthma: Allergic Bronchopulmonary Aspergillosis
Asthma is a disorder of the tracheobronchial tree characterized by inflammation, reversible airway obstruction and tracheobronchial mucosal hyperreactivity to numerous stimuli. Asthma often coexists with other allergic disorders (e.g. allergic rhinitis and atopic dermatitis). Radiography is indicated to exclude other causes of wheezing and to detect complications. Air-trapping due to small airway disease is the most common HRCT feature in children with asthma (Fig. 6) (Fig. 4, “High-Resolution CT of the Lung in Children: Technique, Indications, Anatomy, and Features of Lung Disease”) and can disappear after therapy with bronchodilators (Lucaya et al. 2000) (Fig. 25, “High-Resolution CT of the Lung in Children: Technique, Indications, Anatomy, and Features of Lung Disease”). The sensitivity of HRCT for asthma is likely superior to pulmonary function test; patchy subsegmental involvement can be detected by HRCT even when pulmonary function tests are normal (Sharma et al. 2002). Atelectasis, particularly in the right middle lobe, is also common (Altamirano et al. 1991).
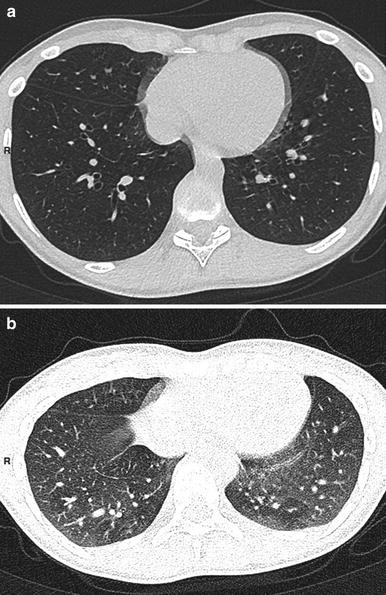
Fig. 6
A 12-year-old boy with severe asthma. Inspiratory scan (a) shows no mosaic pattern. This pattern is clear on expiratory scan (b)
Other reported HRCT features include bronchial wall thickening, bronchiectasis, and mucoid impaction (McLean et al. 1998; Paganin et al. 1992; Grenier et al. 1996). The pathogenesis of bronchial wall thickening in asthmatic patients is not clear. Lynch (1998) has suggested that it is due to inflammation, muscle hypertrophy, and peribronchial fibrosis. The prevalence of bronchiectasis seems to be associated with disease severity (Paganin et al. 1992; Grenier et al. 1996). In contrast with adults with severe asthma, pediatric patients did not have CT evidence bronchiectasis, mucoid impaction, emphysema; but bronchial wall thickening seems to be a criterion of asthma severity in children (Marchac et al. 2002). Jain et al. showed that air trapping on CT scans (percentage of low-density pixels) was correlated with the percentage of predicted total lung capacity and total gas volume and was inversely correlated with the forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) and forced expiratory flow at 25–75 % of forced vital capacity (Jain et al. 2005).
According to Bandeira no isolated HRCT findings (increased lung volume, inspiratory decreased attenuation, expiratory air trapping, and mosaic pattern), could accurately differentiate severe asthma and bronchiolitis obliterans (Bandeira et al. 2011).
Central bronchiectasis associated with asthma is considered to be highly suggestive of allergic bronchopulmonary aspergillosis (ABPA) (Shah et al. 1992; Silva et al. 2004). ABPA is an immunological disorder characterized by immediate hypersensitivity due to endobronchial growth of Aspergillus fumigatus. In patients with IgE-mediated asthma, A. fumigatus may trigger an asthmatic reaction. The diagnosis of ABPA is based on a clinical history of asthma, skin test reactivity, elevated IgE, and measurement of serum precipitins. The presence of randomly distributed, predominantly central, moderate to severe bronchiectasis affecting three or more lobes, bronchial wall thickening, and centrilobular nodules in an asthmatic patient is highly indicative of ABPA (Ward et al. 1999; Mitchell et al. 2000). However, normal HRCT cannot exclude the diagnosis of ABPA (Agarwal et al. 2012).
1.3 Constrictive Bronchiolitis
Constrictive bronchiolitis (bronchiolitis obliterans) is a rare disease characterized by thickening of the bronchiole walls due to submucosal collagenization, with few changes in the distal parenchyma (Colby 1998). Progressive bronchiole narrowing is associated with distortion of the lumen, mucostasis, and chronic inflammation. Bronchiolectasis and bronchiolar smooth-muscle hypertrophy may also be seen.
Constrictive bronchiolitis can be idiopathic or secondary to various insults, such as viral, bacterial, or mycoplasma infections, bone marrow or lung transplantation, collagen vascular diseases or toxic fume inhalation (Chang et al. 1998; Lau et al. 1998; Sargent et al. 1995; Siegel 1999). It has also been reported to occur in association with Stevens-Johnson syndrome (Kim and Lee 1996). Chest X-rays are usually normal, although hyperaeration and vascular attenuation are sometimes seen. HRCT demonstrates a mosaic perfusion pattern due to oligemia and air-trapping, which is better detected on expiratory scans. Central or peripheral bronchiectasis, bronchial thickening, and mucus plugging of the centrilobular bronchioles may also be noted. A recent study using xenon ventilation CT technique demonstrates impaired regional ventilation and its heterogeneity accurately in children with constrictive bronchiolitis (Goo et al. 2010).
Swyer-James or Macleod’s syndrome is a variant of postinfectious constrictive bronchiolitis (Marti-Bonmati et al. 1989), and is characterized by unilateral small or normal-sized hyperlucent lung with air-trapping (Stern and Samples 1992; Moore et al. 1992). It is usually the result of a viral or mycoplasma respiratory infection in early childhood. HRCT reveals unilateral hyperlucency and decreased pulmonary vascularity in all patients. Other common findings are a mosaic perfusion pattern and bronchiectasis, each of which is seen in approximately 70 % of patients. Expiratory HRCT scans show air-trapping in the hyperlucent lung in all cases. Contralateral lung involvement, characterized by patchy areas of air-trapping, is present in half the patients (Fig. 7). Bronchiectasis can be cylindrical or varicose and may be associated with collapse. Children without bronchiectasis or with cylindrical bronchiectasis had a lower incidence of pneumonia episodes than those with varicose bronchiectasis (Lucaya et al. 1998). Several authors reported that the disease might occur in various forms including partial involvement of one lung or bilateral disease (Lucaya et al. 1998; Arslan et al. 2001).
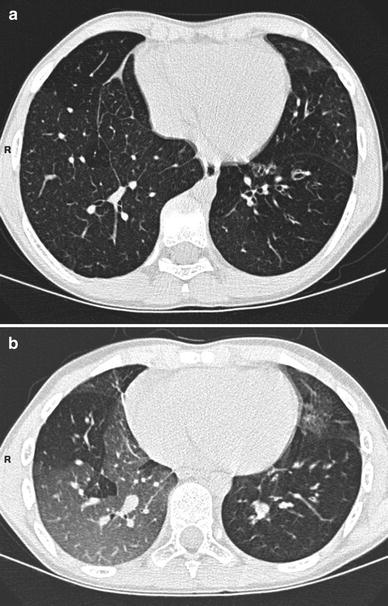
Fig. 7
Inspiratory (a) and expiratory (b) HRCT scans in a 10-year-old boy with postinfectious bronchiolitis obliterans (mycoplasma infection). CT shows bronchiectasis in the lingula and in the left lower lobe. It also demonstrates bilateral mosaic perfusion pattern with air trapping
Constrictive bronchiolitis may occur after heart-lung transplantation (50 % of patients) or bone marrow transplantation (10 % of patients). It is thought to be the consequence of repeated episodes of rejection. Clinically, the patients may present with cough and dyspnea. The triad of mosaic perfusion pattern, bronchial dilatation, and bronchial wall thickening after lung transplantation is indicative of constrictive bronchiolitis (Fig. 8). A mosaic perfusion pattern without the associated bronchial changes has been observed in a large percentage of transplant patients with normal pulmonary function tests (Lau et al. 1998). In lung transplant recipients, the extent of air trapping (in excess of 32 % of the parenchyma) seems to be suggestive of constrictive bronchiolitis (Bankier et al. 2001).
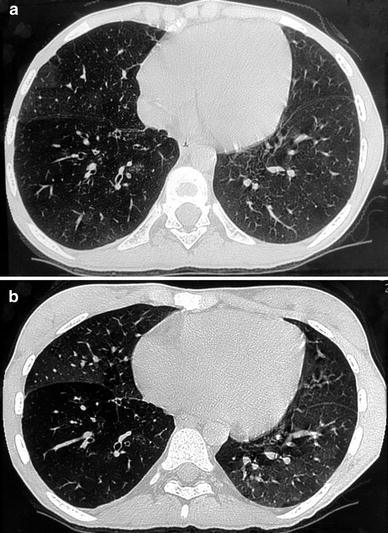
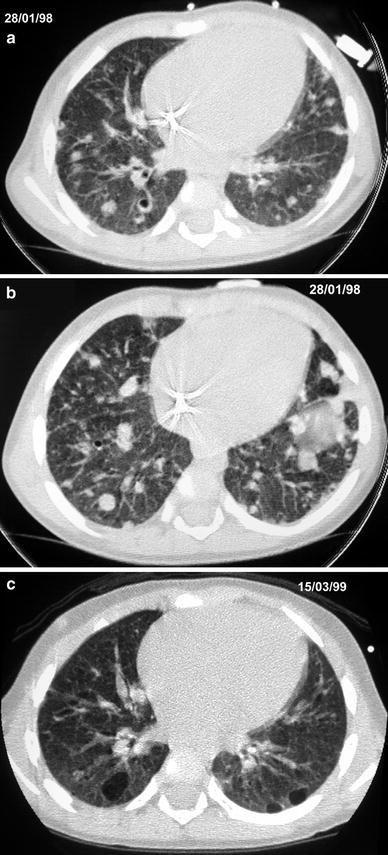
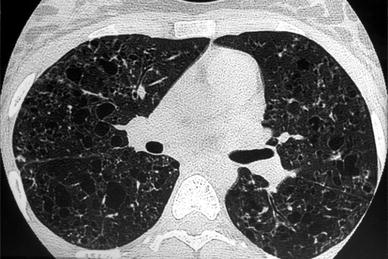
Fig. 8
A 17-year-old girl with constrictive bronchiolitis after heart-lung transplantation. Bronchial dilatation in the right lower lobe with bronchial wall thickening are seen on inspiratory HRCT (a). Mosaic perfusion is better seen on expiratory HRCT (b)
Fig. 9
A 14-month-old girl with Langerhans’ cell histiocytosis. Initial HRCT (a, b) shows nodules and small cystic lesions. Follow-up HRCT (c) at the age of 30 months shows larger cystic lesions
Fig. 10
A 16-year-old girl with Langerhans’ cell histiocytosis. HRCT shows thick- and thin-walled cysts; a few micronodules are also seen
1.4 Bronchiolitis Obliterans Organizing Pneumonia: Organizing Pneumonia
Bronchiolitis obliterans organizing pneumonia (BOOP) is characterized pathologically by the presence of granulation tissue within the lumen of bronchioles and alveolar ducts and associated patchy areas of organizing pneumonia. BOOP rarely occurs in children. It may be idiopathic) but is more commonly seen in children after chemotherapy. It can also occur after bone marrow transplantation or as a response to toxic inhalants, drugs, radiotherapy or viral, mycoplasmal, or bacterial infection (Inoue et al. 1996; Mathew et al. 1994; Kleinau et al. 1997). The main symptoms are cough, dyspnea, fever, and weight loss. Physical examination is unremarkable except for crackles on auscultation of the lungs. Pulmonary function tests show a restrictive ventilatory defect with impaired gas transfer.
The HRCT findings in BOOP most commonly consist of patchy consolidation or ground glass opacities, often with a subpleural and/or peribronchial distribution changing in location over a matter of weeks (Muller et al. 1990; Flowers et al. 1992; Akira et al. 1998; Roberton and Hansell 2011) (Fig. 24, “High-Resolution CT of the Lung in Children: Technique, Indications, Anatomy, and Features of Lung Disease”). The reverse halo sign was initially considered to be an HRCT scan finding characteristic of this disease. This sign was demonstrated in a variety of conditions, including infectious diseases such as tuberculosis, invasive pulmonary disease, pneumocystis jiroveci pneumonia, and noninfectious disease such as pulmonary embolism, sarcoidosis, pulmonary edema, or Wegener disease (Marchiori et al. 2012). Peripheral nodular opacities, irregular linear opacities, bronchial wall thickening and dilatation, and small pleural effusions may also be present (Webb et al. 1996; Lee et al. 1994a). Although nonspecific, the HRCT findings can suggest the diagnosis and help to select the site for biopsy.
In adults, a new classification and terminology is now preferred. The preferred term is organizing pneumonia. Organizing pneumonia is defined pathologically by the presence in the distal airspaces of buds of granulation tissue progressing from fibrin exudates to loose collagen containing fibroblast. The lesions occur predominantly within alveolar spaces but are associated with buds of granulation tissue occupying the bronchiolar lumen (Cordier 2000). The organizing pneumonia, in adult population, is often secondary to a known cause (rheumatoid arthritis, viral pneumonia, dug reactions). The term cryptogenic organizing pneumonia is used when histologic features are demonstrated and the cause is idiopathic (Wittram et al. 2003; Ujita et al. 2004). This new terminology is not widely used in the pediatric literature.
2 HRCT Findings in Specific Diseases
2.1 Chronic Diffuse Infiltrative Lung Disease
A specific diagnosis of chronic diffuse infiltrative lung disease (CDILD) is essential to prescribe treatment. The diagnosis is based on clinical information, pulmonary function tests, bronchoalveolar lavage, and chest imaging. Studies in adults have demonstrated the superiority of HRCT over radiography for obtaining the correct diagnosis of CDILD because many of these patients have distinguishing features (characteristic appearances and distributions) when evaluated with this technique (Grenier et al. 1994; Lee et al. 1994b; Bonelli et al. 1998; Swensen et al. 1997). According to numerous publications the same results were obtained in children (Lynch et al. 1999; Copley et al. 2000; Koh and Hansell 2000). In some cases, lung biopsy can be avoided. HRCT can also be useful to determine the optimal site for biopsy and to assess the extent of the disease. CDILD is uncommon in children. The prevalence of CDILD is estimated between 1.32 and 3.6 per 1,000,000 (Dinwiddie et al. 2002; Griese et al. 2009). During a long period of time, pediatric CDILD description was based on small series. Two main groups were proposed: disorders of known etiology (aspiration, extrinsic allergic alveolitis) and disorders of unknown etiology. In 2004, a task force (Clement et al. 2004) classified CDILD into four groups (interstitial lung diseases of unknown etiology, idiopathic interstitial pneumonias, other forms of interstitial pneumonia and congenital disorders). In 2007, a new classification for infants was proposed by a study group formed by clinicians and pathologists (Deutsch et al. 2007). This classification is divided into four main groups: diffuse developmental disorders, growth abnormalities, surfactant dysfunction disorders, and related abnormalities or specific conditions of unknown or poorly understood etiology (Lee 2013). The understanding of the pathogenesis and diagnosis is due to advances in genetics. Some respiratory diseases in infants (surfactant or some diffuse lung developmental disorders) have a genetic basis (Cazzato et al. 2013). The applications of HRCT are less developed. Our experience suggests that HRCT contributes to the diagnosis and monitoring of pediatric CDILD.
2.1.1 Langerhans’ Cell Histiocytosis
Infiltration and accumulation of monocytes and large histiocytes in various tissues and organs characterize the histological appearance of Langerhans’ Cell Histiocytosis (LCH). Pulmonary involvement is present in 23–50 % of children with the multisystemic form (Smets et al. 1997; Odame et al. 2006). Localized LCH is the mildest and most common form (70 % of all cases) and involves either bone or lung. The lung is the second most common site of LCH (Sminiotopoulos et al. 1999). HRCT detects centrilobular, peribronchial or peribronchiolar granulomas, usually 1–10 mm in diameter; larger nodules are less common. These nodules can disappear or cavitate (Brauner et al. 1989b) and become thick-walled cysts that can progress to thin-walled cysts (Brauner et al. 1997) (Fig. 9).
Thick (wall thickness >2 mm) or thin-walled pulmonary cysts are the main feature of LCH. They may be round or irregularly shaped, probably due to the fusion of several cysts (Fig. 10). LCH lesions are mostly found in the upper and middle lung zones. The costophrenic angles are generally spared in adults (Moore et al. 1989) and involved in children (Seeley et al. 2012). Rupture of subpleural cysts may cause pneumothorax (Fig. 11). Pneumothorax occurs in up to 25 % of patients over the course of their disease (Abbott et al. 2004). In children, LCH lesions may remain stable over long periods or progress rapidly, leading to destruction of the pulmonary parenchyma within a few weeks or months after diagnosis (Seely et al. 1997). In our experience, patients with a poor lung involvement are asymptomatic.
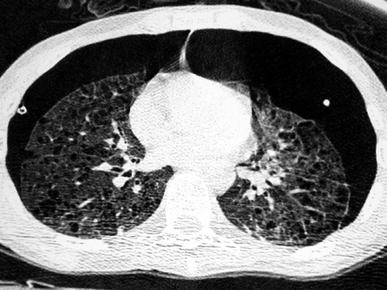
Fig. 11
A 15-year-old boy with Langerhans’ cell histiocytosis, with multiple skin lesions, diabetes insipida, and recurrent bilateral pneumothoraces. Chest CT shows multiple pulmonary cystic lesions, some located subpleurally, and bilateral pneumothorax
Thymic involvement associated with parenchymal lesions has been also reported (Donnelly 2000). Prognosis of primary pulmonary LCH in pediatric age group remains unclear because of limited number of cases (Bano et al. 2014). In the multisystemic form of LCH pulmonary involvement does not mean that the disease is more severe or suggests a poorer prognosis (Smets et al. 1997; Braier et al. 2004), yet it may influence the choice of treatment.
2.1.2 Extrinsic Allergic Alveolitis
Extrinsic allergic alveolitis (EAA) is caused by the repeated inhalation of particulate organic antigens. Farmer’s lung is the best-known EAA syndrome and is a rare entity in young children (Stauffer et al. 2006). The development of EEA requires massive acute or prolonged low-grade exposure. Many inhaled responsible antigens have been described, including animal and plant proteins, and fungal microorganisms (thermophilic actinomycetes). Extrinsic allergic alveolitis is divided into acute, subacute, and chronic forms (Vincent et al. 1992). The diagnosis of EAA in its earliest stages is controversial and remains primarily clinical. Symptoms occur 4–8 h after exposure, and include shortness of breath, dry cough, malaise, and fever. Subacute and chronic forms have an insidious onset with progressive shortness of breath and cough.
HRCT abnormalities are suggestive of the disease and depend on the stage of the disease. HRCT could be useful to evaluate the inflammatory activity of the disease (Sterclova et al. 2006). HRCT could allow the diagnosis in asymptomatic family members of an index case (Ceviz et al. 2006). HRCT is rarely performed in the early stage. In the acute and subacute stages EAA presents as airway disease characterized by small poorly defined centrilobular nodules (<5 mm in diameter) and areas of ground glass (Hansell and Moskovic 1991) (Fig. 12). Ground-glass opacities are slightly more marked in the middle and lower lung zones. Areas of decreased attenuation and air-trapping, consistent with small airway disease, are also common findings (Small et al. 1996). Areas of ground-glass opacities and centrilobular nodules decreased during the follow-up (Tateishi et al. 2011). Chronic EAA is characterized by fibrosis that seems to spare the lung bases.
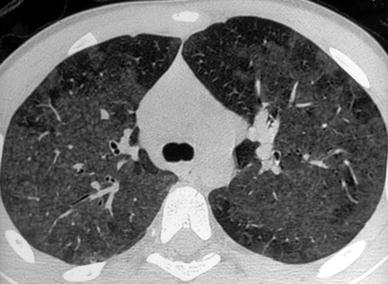
Fig. 12
A 12-year-old boy with extrinsic allergic alveolitis. HRCT shows small, ill-defined rounded opacities with patchy ground-glass opacities
2.1.3 Sarcoidosis
Sarcoidosis is a chronic granulomatous disorder of unknown etiology. It is uncommon in children and occurs most often in young adults (Pattishall and Kendig 1996). The majority of pediatric patients are 9–18 years of age (Grossman et al. 1985). Prognosis is more severe in younger children and in case of multi-organ involvement (Fauroux and Clément 2005) Respiratory symptoms include cough, dyspnea, and, sometimes, chest pain. Mediastinal and/or bilateral hilar adenopathy, often isolated, is the most common intrathoracic finding in sarcoidosis. The characteristic HRCT finding consists in small 2–10 mm nodules with irregular margins distributed along the lymphatics in the bronchovesicular sheath and in the interlobar septa and pleura. This distribution can produce a beaded appearance of the bronchovesicular bundles and interlobular septa and fissural nodularity (Brauner et al. 1989a; Dawson and Muller 1990; Traill et al. 1997) (Fig. 13).
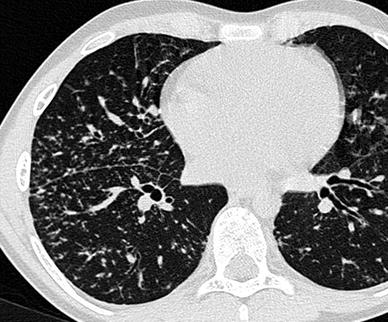
Fig. 13
An 11-year-old boy with sarcoidosis. HRCT detects small nodules in a perilymphatic distribution (note the beaded appearance of the bronchovascular bundles and subpleural nodularity)
Confluence of granulomas may result in large opacities with poorly defined contours, or areas of frank consolidation. Air-bronchograms may be seen within these opacities. Large nodules can cavitate, but this is uncommon. Patchy areas of ground-glass opacity may also be present and may be due to the presence of numerous sarcoid granulomas below the resolution of HRCT (Nishimura et al. 1995). Micronodules and ground-glass opacities are the most frequent features observed in children with sarcoidosis (Sileo et al. 2013).
Ground-glass opacities, architectural distortion, displacement of interlobar fissures, traction bronchiectasis, cystic air spaces, and honeycombing are signs of advanced disease with fibrosis (Abehsera et al. 2000). Posterior displacement of the main or upper lobe bronchus is a classical finding, which indicates loss of volume in the posterior segment of the upper lobes.
HRCT could assess the severity of the disease. The appearance and the extent of the disease on HRCT (thickening of the bronchovesicular bundle, intraparenchymal nodules, septa and nonseptal lines, and focal pleural thickening are associated with parameters of respiratory functional impairment (Drent et al. 2003).
2.1.4 Pulmonary Alveolar Proteinosis
Pulmonary Alveolar Proteinosis (PAP) is a rare intrinsic lung disease characterized by alveolar filling with amorphous lipoproteinaceous material (Schumacher et al. 1989). The cause of alveolar proteinosis is unclear. One possible cause may be dysfunction of intra-alveolar macrophages, another cause relates to an abnormal surfactant C protein function which predisposes certain patients to diffuse lung disease (Nowers et al. 2002). Plain films show alveolar infiltrates, a reticulonodular pattern or both. Pleural effusion and adenopathy are absent (McCook et al. 1981). HRCT shows areas of consolidation or ground glass, often with a geographic distribution, and/or widespread miliary nodules (Godwin et al. 1988; Murch and Carr 1989; Albafouille et al. 1999) (Fig. 14). Smooth thickening of the interlobular septa within the areas of air space disease resulting in a crazy paving appearance (Fig. 45, “High-Resolution CT of the Lung in Children: Technique, Indications, Anatomy, and Features of Lung Disease”) is suggestive of, but not specific to, alveolar proteinosis (Coullier et al. 1999; Johkoh et al. 1999b; Rossi et al. 2003). Quantitative CT is proposed for detecting the response to therapeutic interventions such as whole lung lavage (Guan et al. 2012).
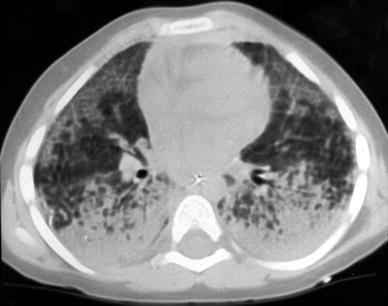
Fig. 14
A 3-year-old boy with biopsy-proven pulmonary alveolar proteinosis. Despite the 5 mm slice thickness alveolar consolidation and alveolar infiltrates are clearly visualized
2.1.5 Pulmonary Fibrosis and Chronic Interstitial Pneumonias
Pulmonary fibrosis is a chronic inflammatory interstitial lung disorder, characterized by an initial accumulation of inflammatory and immunoregulatory cells in the pulmonary interstitium and the alveolar space. Inflammation leads to modification of the alveolar structures with progression to interstitial fibrosis and thickening of alveolar walls. In children, pulmonary fibrosis is the result of a heterogeneous group of disorders that share common histological features (Osika et al. 1997). The known causes include infectious disorders, reactions to environmental exposures, drugs, collagen-vascular disorders, or gastroesophageal reflux with chronic aspiration. For idiopathic pulmonary fibrosis (IPF), classification by histological features into usual interstitial pneumonitis (UIP) and desquamative interstitial pneumonitis (DIP) has been proposed. The distinction between these two forms of fibrosing alveolitis is now questioned (UIP and DIP can be seen simultaneously). These entities may represent different stages of a lung injury (Webb et al. 1996) with associated thickening of alveolar walls and many mononuclear cells in the alveolar space.
Because of the rarity of these entities in children, most HRCT findings have been reported in adults. On HRCT, the main sign in DIP is the presence of bilateral and symmetric areas of ground-glass opacity (the predominant lesion in DIP is alveolar spaces filled with macrophages) (Hartman et al. 1993). The ground-glass areas of attenuation are seen mainly in lower lung zones. Other findings in DIP are those of UIP: reticular opacities, which correspond to areas of irregular fibrosis, honeycombing, and traction bronchiectasis (Nishimura et al. 1992). Less common HRCT findings include discrete nodules and interlobular septal thickening. Mild enlargement of mediastinal lymph nodes is commonly seen, whereas large lymph nodes are uncommon.
The main HRCT findings of IPF in children are areas of ground-glass attenuation involving mostly the subpleural regions Seely et al. (1997). Large subpleural air cysts in the upper lobes adjacent to areas of ground-glass opacities seem to be unique to childhood IPF. These cysts are interpreted as paraseptal or irregular emphysema (Fig. 15). Intralobular lines, irregular interlobular septal thickening and honeycombing seem to be less common findings and, thus, less contributive to diagnosis.
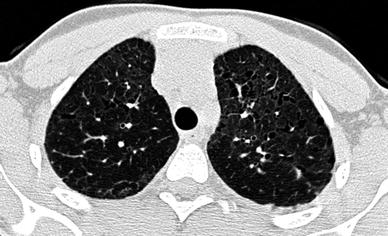
Fig. 15
A 13-year-old boy with biopsy-proven idiopathic pulmonary fibrosis. HRCT shows ground-glass attenuation, intralobular lines, and air cysts and involving mostly the subpleural regions
Recently, two new forms of idiopathic interstitial pneumonia have been described: acute interstitial pneumonia (AIP) (Katzenstein et al. 1986) and nonspecific interstitial pneumonia and fibrosis (NIPF) (Katzenstein and Fiorelli 1994).
AIP is a fulminant disease of unknown etiology that is histologically characterized as diffuse alveolar damage. The latter manifests as injury to the alveolar lining and endothelial cells, pulmonary edema, hyaline membrane formation and, later, proliferative changes involving alveolar and bronchiolar lining cells, and interstitial cells. The histologic appearance of AIP can be separated into acute exudative, subacute proliferative and chronic fibrotic phases. The radiological finding on chest radiographs is progressive parenchymal consolidation. HRCT images include diffuse air-space consolidation and patchy or diffuse ground-glass opacities, traction bronchiectasis, and, occasionally, focal honeycombing (Primack et al. 1993; Ichikado et al. 1997). These findings are usually bilateral, symmetrical, and basilar in distribution. Ground-glass opacities are seen in all three histological phases and reflect different histological findings. During the acute exudative phase, they reflect the presence of alveolar septal edema and hyaline membranes along the alveolar walls. During the subacute proliferative phase, ground-glass opacities are due to intra-alveolar and interstitial organization. During the fibrotic phase, ground-glass attenuation results from alveolar septal fibrosis. Bronchiectasis within areas of ground-glass attenuation may correspond to fibrosis and its severity (Johkoh et al. 1999a, c).
NIPF describes the group of interstitial pneumonias that cannot be classified as UIP, DIP, AIP, or BOOP. NIPF is essentially a diagnosis of exclusion. It is characterized by varying degrees of interstitial inflammation and fibrosis that persist (Müller and Colby 1997). Copley et al. (2000) reported six cases of NIPF in children. In three of them, HRCT showed a predominantly upper-zone honeycomb pattern with parenchymal distortion superimposed on a background of widespread ground-glass opacification (Fig. 16). For the other three patients with NIPF, one had widespread ground-glass opacification, honeycombing with mid- and lower-zone predominance, and traction bronchiectasis; another had widespread ground-glass opacification; and the last patient had widespread ground-glass opacification with peripheral consolidation. None of the patients had interlobular septal thickening.
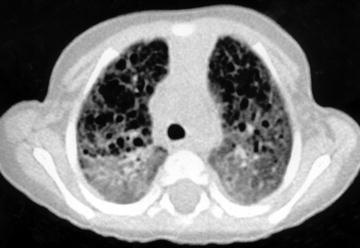
Fig. 16
A 10-month-old female infant with biopsy-proven nonspecific interstitial pneumonitis. HRCT shows ground glass and honeycombing. [Reprinted with permission from Copley et al. (2000)]
Genetic studies will perhaps help us to better understand these entities in children. Recently, mutations in the gene encoding surfactant protein C associated with familial interstitial lung disease have been identified in familial cases (Nogee et al. 2001). Few authors report familial cases of NIPF and UIP with mutation in surfactant protein C (Thomas et al. 2002; Chibbar et al. 2004).
2.1.6 Lymphocytic Interstitial Pneumonia
LIP is a benign lymphoproliferaltive disorder described by Liebow and Carrington (1973) and characterized by pulmonary infiltration of lymphocytes and plasma cells. LIP occurs in patients who have systemic disorders, such as Sjögren’s syndrome, multicentric Castleman’s disease or acquired immunodeficiency syndrome (AIDS). In children, LIP has been reported to be frequently associated with AIDS. In a series of 77 human immunodeficiency virus-positive (HIV+) children evaluated by Amorosa et al. (1992), 32 were diagnosed as having LIP. The appearance of LIP on chest radiography is nonspecific and includes a fine reticular pattern and nodular opacities or a diffuse confluent pattern. HRCT findings include extensive bilateral ground-glass attenuation, focal air-space consolidation, ill-defined, micronodules with a perilymphatic distribution, and thin-walled cystic lesions (Carignan et al. 1995; McGuinness and Naidich 1995; Becciolini et al. 2001) (Fig. 17). Associated bronchiectasis and hilar or mediastinal lymphadenopathy may be present (Johkoh et al. 1999b). No pleural effusion is seen in LIP (Honda 1999).
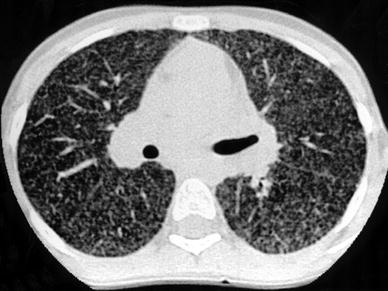
Fig. 17
A 13-year-old girl with biopsy-proven lymphocytic interstitial pneumonia. The patient was immunocompromised because of postviral neutropenia. HRCT shows profuse nodules with random distribution. [Reprinted with permission from Copley et al. (2000)]
2.1.7 Pulmonary Lymphangitic Carcinomatosis
Pulmonary lymphangitic carcinomatosis (PLC) refers to tumor growth in the lymphatics of the lung. The histological findings are characterized by thickening of the interlobular septa and the peribronchovascular interstitium. In most cases, the primary tumor disseminates hematogenously to the lungs and secondarily penetrates vessel walls and invades the surrounding interstitium and lymphatics. In children PLC is uncommon but can occur in lymphoma, thyroid carcinoma, sarcoma, and neuroblastoma (Kuhn 1993). In our experience, PLC was most common in lymphoma.
Chest X-ray findings are normal or show nonspecific findings in many patients with PLC (Munk et al. 1988). HRCT findings (Johkoh et al. 1992) correlate well with the two types of lymphatic drainage systems described by pathologists. Axial drainage is seen on HRCT as smooth or nodular peribronchovascular interstitial thickening in the parahilar lung and enhanced visibility of the branching arteries in the pulmonary lobule. Peripheral drainage (interlobular and subpleural) is seen as interlobular septal thickening or as thickening of fissures, which may be smooth or nodular. HRCT abnormalities can be focal, unilateral, or diffuse (Fig. 22, “High-Resolution CT of the Lung in Children: Technique, Indications, Anatomy, and Features of Lung Disease”) (Fig. 18). Despite axial and peripheral interstitial abnormalities, lung architecture remains normal, a finding that is useful to differentiate between PLC and sarcoidosis.
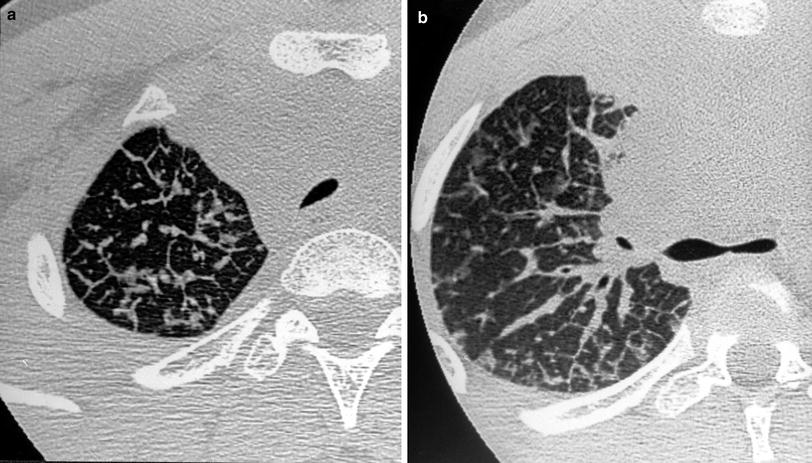
Fig. 18
A 14-year-old boy with Hodgkin’s disease. HRCT shows thickening of the interlobular septa (a), smooth peribronchovascular interstitial thickening (b), and ground-glass attenuation
2.1.8 Collagen-Vascular Disease and Pulmonary Vasculitis
Pulmonary vasculitis can be due to primary systemic vasculitides, such as Wegener’s granulomatosis, Churg-Strauss angiitis, or microscopic polyangiitis. In addition, pulmonary vasculitis may accompany systemic connective tissue disease, including systemic lupus erythematosus, dermatomyositis, or systemic sclerosis. Connolly et al. (1996) identified a pattern on HRCT of perivascular, centrilobular, ill-defined densities in eight children with vasculitis. In the appropriate clinical setting this pattern indicates pulmonary involvement and may obviate the need for lung biopsy.
Collagen-vascular disease, especially progressive systemic sclerosis (PSS), is commonly associated with pulmonary fibrosis in children. Seely et al. (1998) described a series of 11 patients with PSS who had interstitial lung disease. HRCT revealed abnormality in 91 % of the patients. The main features were ground-glass opacities, subpleural micronodules, nonseptal linear opacities, honeycombing, and subpleural cysts. HRCT was able to demonstrate interstitial lung disease in 53 % of patients with lupus erythematosus (Fenlon et al. 1996) (Fig. 19). In the pediatric population, the frequency of pulmonary involvement is lower (8 %) than in adult one (Lilleby et al. 2006). Juvenile rheumatoid arthritis, on the other hand, seldom leads to pulmonary fibrosis in children (Seely et al. 1998). The HRCT pattern of fibrosis associated with collagen-vascular disease is similar to that of idiopathic pulmonary fibrosis and consists predominantly of areas of ground-glass involving mostly the subpleural lung regions, large and thin-walled cysts or bullae in the affected upper-lung zones, and smooth or irregular intralobular septal thickening.
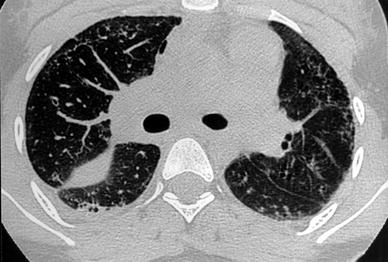
Fig. 19
A 16-year-old girl with systemic lupus erythematous. HRCT shows honeycombing, predominantly in the periphery, and pleural effusion. Note the right pneumomediastinum
2.1.9 Pulmonary Lymphangiectasia
According to Noonan et al. (1970), pulmonary lymphangiectasia can be divided into three groups. In the first, pulmonary lymphangiectasia is part of a generalized disease, the major clinical manifestations being related to the intestinal involvement. The pulmonary involvement is less severe and is associated with a better prognosis than for the following two groups. In the second group with associated heart disease, dilatation of the lung lymphatics occurs secondary to obstruction of pulmonary venous flow. The third group, termed congenital pulmonary lymphangiectasia (CPL), includes patients with a primary developmental defect of lung lymphatics, which are dilated. Histological examination is characterized by subpleural, interlobar, perivascular, and peribronchial lymphatic dilatation. Radiological findings include bilateral pulmonary hyperinflation and a reticulonodular pattern throughout the lung fields. Occasional small cystic areas, representing aerated distal bronchial and alveolar ducts, may also be present. Pleural effusion and pneumothorax may be associated. Unilateral or lobar involvement has been reported (Verlaat et al. 1994; Li et al. 1985; Rettwitz-Volk et al. 1999).
Prolonged survival of patients with CPL is rare. The chest radiograph and HRCT findings in survivors have recently been reviewed by Chung et al. (1999). Chest radiograph findings include increased interstitial markings, hyperinflation that generally increases with age, pleural effusion, and pectus excavatum. The presence of patchy subpleural or perihilar ground-glass opacities that are fixed in location and tend to decrease with time was the most characteristic HRCT feature. Hyperinflation and interstitial thickening were often seen.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


