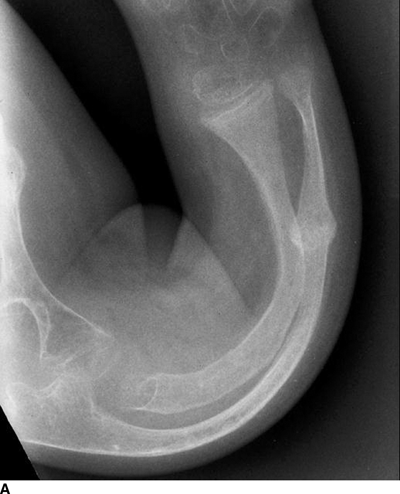
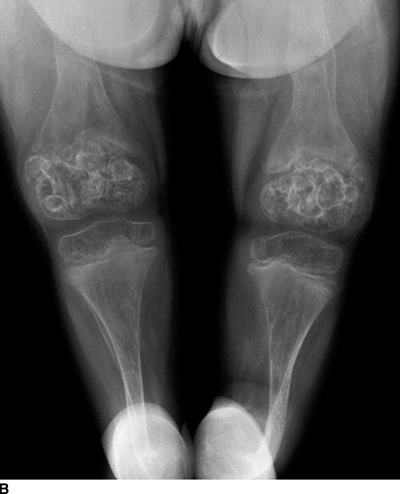
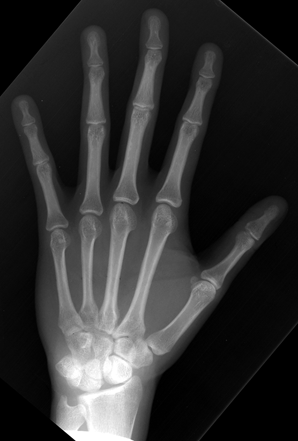
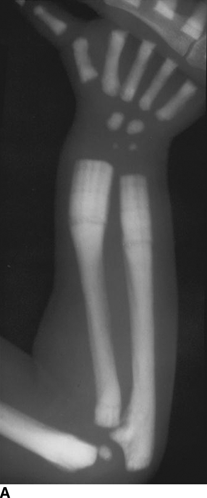
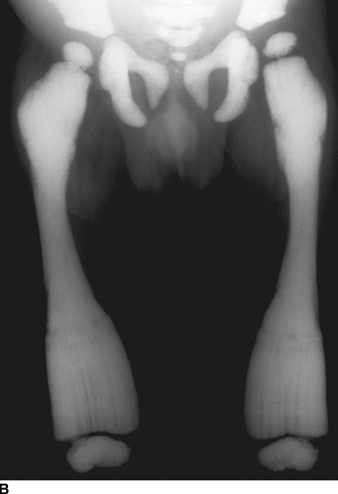
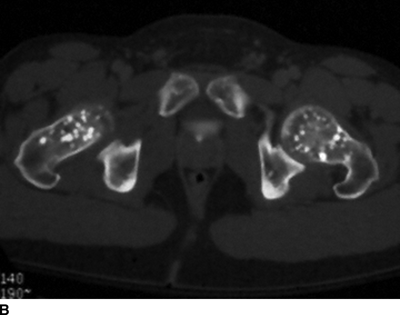
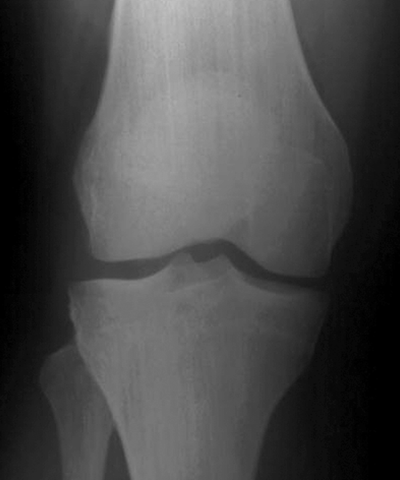
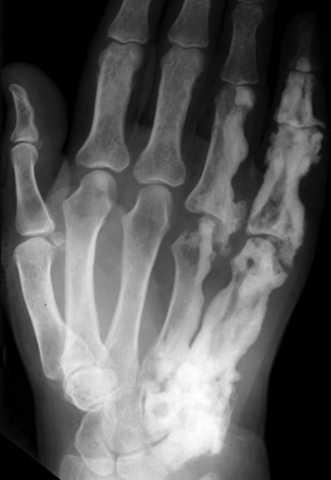
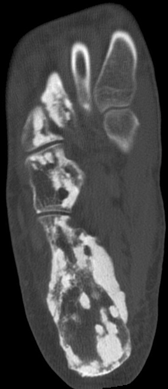
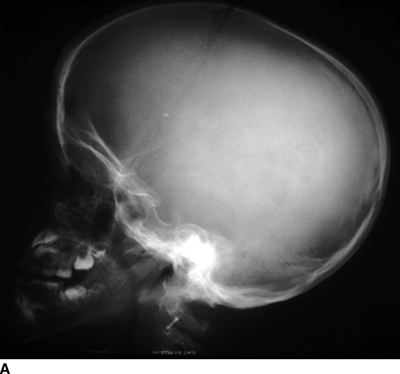
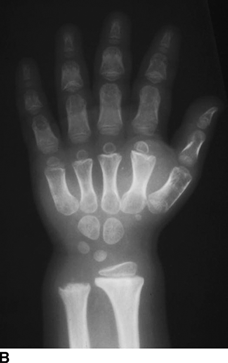
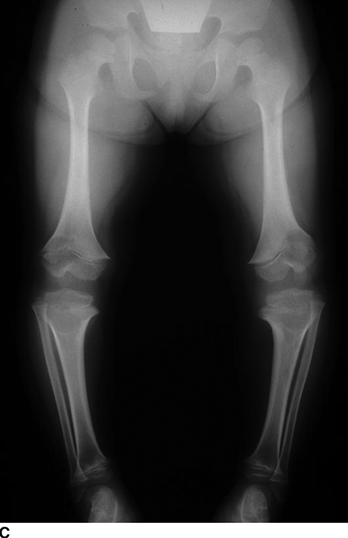
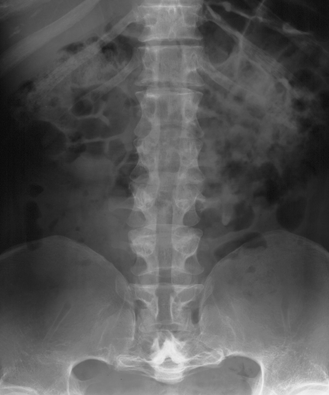
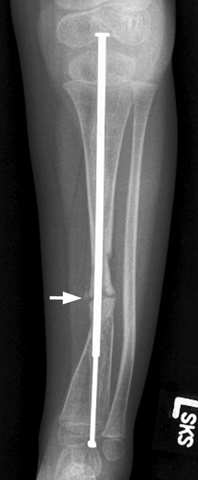
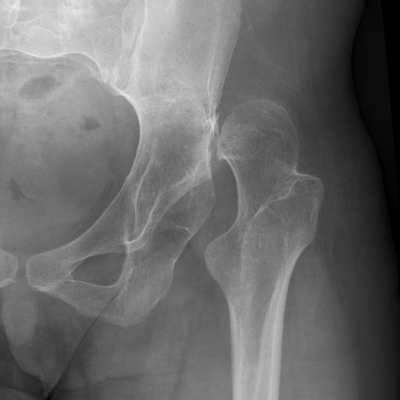
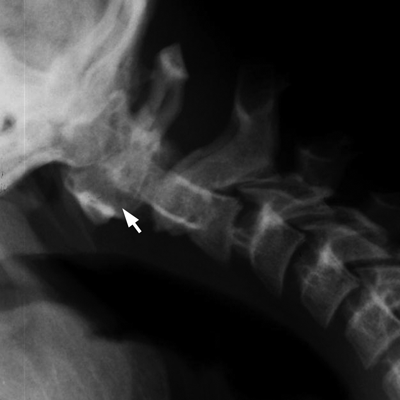
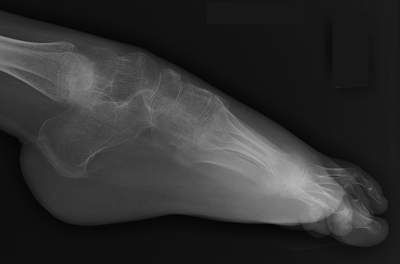
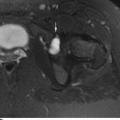
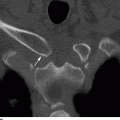
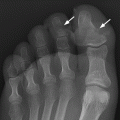
CHAPTER 14 This chapter describes the radiology of many developmental and congenital conditions that affect the musculoskeletal system. Some of these conditions may not present until adolescence or adulthood. The skeleton develops and matures with a consistent and irreversible sequence of changes in osseous size and contour. Particular features in the contours of developing bones are useful maturational landmarks in the sequence of ossification. These features are used to estimate skeletal age. The most practical method is to use the radiographic atlas of Greulich and Pyle. This atlas contains radiographs of the left hand and wrist in boys and girls of different ages—correlated with their chronologic age—from the neonate to the skeletally mature. A radiograph of the left hand and wrist of the individual patient is compared to these standards, and a best match is found. Each chronologic age has a mean bone age and a standard deviation in bone age. The radiologic report of the bone age should include the bone age itself as well as the standard deviation in the bone age for the patient’s chronologic age. Greulich and Pyle’s atlas is based on radiographs of healthy children of northern European ancestry from Cleveland, Ohio, whose families were incidentally above average in economic and educational status. These children were followed between 1931 and 1942 as part of a longitudinal long-term investigation of human growth and development. This project produced atlases of skeletal development of the hand and wrist, the knee, and the foot and ankle. The applicability of these atlases to individuals from other ethnic and socioeconomic populations is uncertain. Osteogenesis imperfecta is a group of inborn connective tissue disorders characterized by radiographically decreased bone density. The underlying problem is one of abnormal collagen synthesis, in which a variety of different molecular defects in collagen produces a continuous spectrum of phenotypes. In the skeleton, the bone matrix is deficient, resulting in thin, osteoporotic, and fragile bones that are subject to repeated insufficiency fractures and consequent deformity (Fig. 14.1). The condition is heritable, but cases are often sporadic. In general, there are autosomal-recessive severe forms that present at birth and autosomal-dominant forms that present later and have a mild course. The condition ranges from severe, congenital involvement with multiple fractures in utero and perinatal death to mild, late manifestations in adulthood. The severe forms account for 10% of cases; the less severe forms account for the remaining 90%. The incidence of osteogenesis imperfecta is 1 per 20,000 to 60,000 live births. Associated clinical features—with variable expression— include blue sclerae (90%); thin, translucent skin; hypermobile, lax peripheral joints; abnormal teeth (dentinogenesis imperfecta); and deafness (fragile otic bones). FIGURE 14.1. Osteogenesis imperfecta. A: Radiograph of forearm shows osteoporosis, bowing deformity, and healing insufficiency fractures of different ages. B: Lower extremities show similar findings along with popcorn epiphyses. Marfan syndrome, homocystinuria, and Ehlers-Danlos syndrome are heterogeneous groups of heritable connective tissue disorders with musculoskeletal manifestations. Marfan syndrome is associated with excessive height, long spidery digits (arachnodactyly), hypermobile joints, and scoliosis (Fig. 14.2). Ocular and cardiovascular abnormalities dominate the clinical picture. Accumulation of amino acid metabolites in homocystinuria interferes with collagen cross linking, leading to fragile bones, scoliosis, and excessive length of long bones. Ehlers-Danlos syndrome is associated with fragile skin and hypermobile joints. FIGURE 14.2. Marfan syndrome. Radiograph of the hand shows arachnodactyly. Hypermobility is present at the carpus, with overlap of the proximal and distal carpal rows. The sclerosing skeletal dysplasias are a heterogeneous group of conditions characterized radiographically by dense bones. Osteopetrosis is a group of heritable diseases characterized by reduced bone resorption from osteoclast failure. Many genetic defects may cause osteoclast failure, each having different underlying biochemical and histopathologic abnormalities. One form is associated with renal tubular acidosis and rickets. The final common effect is that bone remodels incompletely or not at all. There are three major clinical groups: infantile-malignant autosomal recessive, which is fatal within the first few years of life (in the absence of effective therapy); intermediate autosomal recessive, which appears during the first decade of life but does not follow a malignant course; and autosomal dominant (Albers-Schönberg disease or marble bones), which has a normal life expectancy but many orthopedic problems. The infantile variant has clinical manifestations that correlate with the lack of marrow development (anemia and thrombocytopenia) and the lack of enlargement of bony remodeling (small cranial foramina result in cranial nerve dysfunction, hydrocephalus, convulsions, and mental retardation). Radiographs show uniformly dense bones with no medullary space, broadened metaphyses (Erlenmeyer flask deformity), and bone-within-a-bone appearance at the tarsals, carpals, phalanges, vertebrae, and iliac wings (Fig. 14.3). Transverse insufficiency fractures occur often. Medical treatments involve high-dose calcitriol to stimulate osteoclast differentiation and bone marrow transplantation to provide monocytic osteoclast precursors. The intermediate and autosomal-dominant forms have mild manifestations that include anemia, cranial nerve palsies, and orthopedic problems. The fragility of osteopetrotic bone and its inability to remodel in response to stress result in repeated insufficiency fractures. Coxa vara, long-bone bowing, hip and knee degenerative arthritis, and mandibular and long-bone osteomyelitis may also occur. Even milder forms of osteopetrosis may be asymptomatic and discovered incidentally on radiographs (Fig. 14.4). FIGURE 14.3. Osteopetrosis, autosomal recessive type (infantile). A: Radiograph of the forearm shows dense bones lacking a medullary space. B: Radiograph of femurs shows club-shaped metaphyses. FIGURE 14.4. Osteopetrosis, incidental adult presentation. Note the sandwich appearance of the vertebrae. Osteopoikilosis (spotted bones, osteopathia condensans disseminata) is an uncommon osteosclerotic dysplasia with sporadic and familial occurrence. Symptoms are generally absent or mild, and its discovery is frequently incidental. The condition is defined by the presence of numerous small, well-defined, homogeneous ovoid or circular foci of increased radiodensity clustered in periarticular osseous regions. The distribution is symmetric, typically involving the epiphyses and metaphyses of long bones, as well as the carpus, tarsus, scapula, and pelvis (Fig. 14.5). The lesions may increase or decrease in size or number. They are histologically identical to bone islands but have no clinical significance unless mistaken for other lesions. FIGURE 14.5. Osteopoikilosis. A: AP radiograph. B: CT scan. Osteopathia striata (Voorhoeve disease) is a rare condition whose transmission is probably autosomal dominant. The condition is defined by the presence of linear, regular bands of increased radiodensity extending from the metaphyses to the diaphyses of tubular bones and the pelvis (Fig. 14.6). Osteopathia striata is usually bilateral, and it is unusual for it to involve the small bones of hands or feet, skull or face, or spine. The cause and pathogenesis are unknown. Clinical manifestations are usually absent, although facial deformity has been described. Discovery is usually incidental. FIGURE 14.6. Osteopathia striata. Melorheostosis is a rare, nonhereditary condition that presents in childhood with extremity pain, limitation of motion, and intermittent joint swelling. Growth disturbances, soft-tissue involvement, and increasing muscle contractures due to tendon and ligament shortening may result in considerable deformity and disability. The condition is commonly limited to a single limb, usually lower. Radiologically, there is extensive periosteal or endosteal cortical hyperostosis with bony excrescences extending along the length of the bone, giving it a wavy contour that has been likened to wax dripping down a candle (Figs. 14.7 and 14.8). Endosteal hyperostosis may fill the medullary cavity, but periosteal and endosteal hyperostosis usually do not occur together. The radiologic appearance is characteristic. The cause and pathogenesis are unknown; the bony excrescences are histologically normal bone. FIGURE 14.7. Melorheostosis involving the fourth and fifth rays of the hand. FIGURE 14.8. Melorheostosis involving the foot shown on axial CT scan. Dwarfism is the condition of being disproportionately undersized. Skeletal dysplasias with dwarfism are legion. Dozens of specific types are listed in the International Nomenclature of Constitutional Diseases of Bone as revised in 1983 by the European Society of Pediatric Radiology. For most of these conditions, there are only a handful of reported cases. Many conditions are lethal, none are reversible, and most are not defined on a radiologic basis. The distinction among the dysplasias with dwarfism often requires detailed genetic, biochemical, histochemical, and histopathologic analysis. Achondroplasia is the most common type of dwarfism and has a classic radiologic appearance. Achondroplasia is the result of a generalized defect in enchondral bone formation, leading to underdevelopment of the portions of bones that grow by this mechanism. The result is a symmetric short-limbed dwarfism in which the proximal segments of the extremities are disproportionately short (rhizomelic micromelia). Because periosteal bone growth is unaffected, the shafts of the long bones are of normal diameter. The fingers are short and stubby. The skull base, formed by enchondral bone formation, is abnormally short and has a small foramen magnum. The calvaria, formed by intramembranous bone formation, is appropriately large for the intracranial contents, giving the head a characteristic brachycephaly with frontal bossing and a small face (Fig. 14.9). The spinal canal is narrow in both anteroposterior and lateral dimensions, but the trunk length is nearly normal. There is an exaggerated lumbar lordosis with prominent buttocks. These abnormalities are usually evident at birth and become more apparent with age. Achondroplasia has autosomal-dominant genetic transmission, but most cases are sporadic. The classic form is heterozygous, has no associated congenital defects, and is compatible with a normal life span. Complications of congenital spinal stenosis are common in adulthood (Fig. 14.10). The homozygous condition is lethal in infancy and has a radiologic appearance identical to thanatophoric dwarfism. Thanatophoric (“death-bringing”) dwarfism is the most common lethal bone dysplasia. FIGURE 14.9. Achondroplasia. A: The base of the skull is short and the face is small. B: The bones are disproportionately short and the fingers appear stubby. C: The femurs are short and thick. FIGURE 14.10. Achondroplasia in adult with spinal stenosis in the lumbar region. Laminectomy has been performed at all levels. Other types of dwarfism may be characterized and classified by the predominant location of disproportionate shortening. Mesomelic micromelia refers to shortening of the forearms and lower legs. Examples of mesomelic dwarfism include camptomelic dwarfism and dyschondrosteosis of the Nievergelt and Langer types. Acromelic micromelia refers to shortening of the hands and feet. Examples of acromelic dwarfism include asphyxiating thoracic dystrophy (Jeune syndrome), chondroectodermal dysplasia (Ellis-van Creveld syndrome), and pyknodysostosis. Many of these dysplasias are amenable to prenatal diagnosis with obstetric sonography, reducing even further the role of radiography. Neurofibromatosis is one of the heritable phakomatoses, characterized by multisystem involvement of hamartomas involving all three germ cell layers. Neurofibromatosis has autosomal-dominant transmission. There are multiple subtypes. Major musculoskeletal manifestations include scoliosis, mesodermal dysplasia, and neurofibromas (Fig. 14.11). Although the scoliosis may be gently curving and identical to idiopathic scoliosis, occasionally a sharply angulated dysplastic thoracic kyphoscoliosis is present and virtually diagnostic. Mesodermal dysplasia may result in focally abnormal or deficient bone formation. Orthopedic complications in neurofibromatosis include bowing, pathologic fracture, pseudoarthrosis, and defective fracture healing (Fig. 14.12). The tibia is a characteristic site for pseudoarthrosis; repeated failures to heal after treatment often lead to amputation. Neurofibromas may erode adjacent bones and present as soft-tissue masses. FIGURE 14.11. Neurofibromatosis in an adult. A: Axial T1 MRI shows multiple lobulated soft-tissue mass in and around the right buttock with dysplastic underlying bone and muscle atrophy. B: Axial T2 MRI shows high signal within the masses. FIGURE 14.12. Neurofibromatosis with pseudoarthrosis (arrow) of the tibia in a child. A telescoping rod has been placed for mechanical support. Cerebral palsy can result in generalized or focal neuromuscular disease in which secondary bone changes and complications are seen. These changes are the result of abnormal stresses on the growing skeleton from muscular spasm, weakness, atrophy, and imbalance. When the normal skeleton remodels in response to abnormal stress, dysplastic changes in the bones and joints may result. Long bones often appear overtubulated (narrow shaft, broad epiphysis). Hip dysplasia secondary to chronic, spastic dislocation is common. Spasm of the thigh adductor muscles positions the femoral head on the lateral acetabular rim, resulting in deformity, subluxation, and, ultimately, hip dislocation (Fig. 14.13). Scoliosis, soft-tissue atrophy, and flexion or extension contractures may be found. Foot deformities of various kinds are common. Similar developmental changes are seen in conditions such as arthrogryposis, muscular dystrophy, peripheral nerve palsy, and polio. FIGURE 14.13. Adult with DDH resulting from cerebral palsy and chronic hip dislocation without weight-bearing. Muscular dystrophy comprises a heterogenous group of genetic diseases characterized by progressive atrophy of skeletal muscle. Depending on the particular genetic defect and its expression, age of onset, anatomic distribution, severity, rate of progression, and pattern of inheritance vary among the different types of muscular dystrophy, dozens of which have been described. Radiologic features of muscular dystrophy in the musculoskeletal system include fatty replacement of muscles, usually symmetric and diffuse (Fig. 14.14), and, if the onset is during skeletal maturation, dysplastic bone changes. Multisystem manifestations may also be present in some forms of muscular dystrophy. FIGURE 14.14. Limb girdle muscular dystrophy in an adult. Oblique coronal T1-weighted MRI shows fatty replacement of all of the shoulder muscles. Arthrogryposis multiplex congenita is an uncommon nonhereditary congenital disorder that manifests with deformed joints, dislocations, and muscle wasting (Fig. 14.15). The deformities are widespread and bilateral and usually cause severe disabilities, but the patient’s life span is often normal. Frequent deformities include clubfoot (talipes equinovarus, present in 75% of cases), flexion deformity of the knee (60%), flexion deformity of the hip (40%), and dislocation of the hip with resultant dysplasia (40%). FIGURE 14.15. Arthrogryposis with poor muscle development and recurvatum deformity (anterior to left; arrow points to cartilaginous patella). Generalized ligamentous laxity in Down syndrome (also called trisomy 21) may be manifested by atlantoaxial (C1–2) subluxation (Fig. 14.16), recurrent patellar dislocation, pes planus (flatfoot deformity), and voluntary, painless hip dislocation. The iliac wings may have a flared appearance. The significance of atlantoaxial subluxation with respect to participation in sports is controversial. FIGURE 14.16. Down syndrome with atlantoaxial subluxation (arrow). Charcot-Marie-Tooth disease (also called hereditary motor and sensory neuropathy, hereditary sensorimotor neuropathy, or peroneal muscular atrophy) is a collection of heritable neuropathies that present with predominant involvement of the lower extremities in late childhood or early adulthood. Foot drop, cavus foot (Fig. 14.17), hammertoe deformities, and muscle atrophy in the peroneal nerve distribution, and scoliosis are common features, but the upper extremities may also be involved. The multiple types of this disease are distinguishable by specific chromosomal abnormalities. FIGURE 14.17. Charcot-Marie-Tooth disease in a young adult with cavus foot, hammertoe deformities, and muscle atrophy. Multiple epiphyseal dysplasia (dysplasia epiphysealis multiplex, Fairbank disease) is the most common of a heterogeneous group of disorders characterized by abnormal growth and development of epiphyses at multiple sites. Multiple epiphyseal dysplasia has autosomal-dominant transmission. The primary defect involves an abnormality in the biochemical composition of the ground substance produced by the epiphyseal chondrocytes, resulting in delayed and disorderly ossification, joint incongruity, and secondary degenerative change. After onset in early childhood, the variable clinical manifestations include articular pain, gait disturbances, short stature, and involvement of multiple sites. Radiographically, the secondary ossification centers occur late and are irregular in morphology. After skeletal maturity, the epiphyses remain irregular and abnormal in shape, tending to be flattened and squared off (Fig. 14.18). Early osteoarthritis is a common sequela. FIGURE 14.18. Multiple epiphyseal dysplasia (dysplasia epiphysealis multiplex). The cardinal features of cleidocranial dysplasia (cleidocranial dysostosis) are absence of a portion of the clavicles and poor ossification of the skull with wormian bones and hypoplastic paranasal and mastoid sinuses. Sometimes, there may be total absence of the clavicles (Fig. 14.19). Other dysplastic features may be present throughout the skeleton, including hypoplastic limb girdles, dysplastic long bones with narrow diaphyses and expanded ends, and spina bifida occulta at multiple levels. Cleidocranial dysplasia is an autosomal-dominant condition with high penetrance. FIGURE 14.19. Cleidocranial dysplasia. AP Radiograph obtained because of trauma incidentally shows complete absence of both clavicles. Radioulnar synostosis is an anomaly of longitudinal segmentation in which the proximal radioulnar joint space fails to form (Fig. 14.20). It occurs both sporadically and familially and is bilateral in approximately 60% of cases. It may also be seen in a variety of other developmental syndromes and can be acquired from conditions such as infantile cortical hyperostosis or infection. Patients have functional impairment of ability to pronate and supinate the forearm. FIGURE 14.20. Radioulnar synostosis. Proximal focal femoral deficiency is a spectrum of bony deficiencies of the proximal femur, ranging from shortening and varus deformity of the shaft to aplasia of the femoral head, neck, and proximal shaft. Dysplastic changes in the acetabulum correlate with the degree of femoral head deformity or hypoplasia. It is congenital but not heritable; the cause is unknown. The condition can be bilateral (Fig. 14.21). FIGURE 14.21. Bilateral proximal focal femoral deficiency. Tibia vara (also called Blount disease) is a growth disorder of the medial portion of the proximal tibial growth plate that results in a varus deformity. The causes of tibia vara are thought to be multifactorial, but the end result appears to be an injury to the medial growth plate, slowing its growth, leading to the varus deformity from continued normal growth on the lateral side of the growth plate. On radiographs, beaking, irregularity, and distortion of the metaphysis are seen along with the varus deformity at the growth plate (Fig. 14.22). FIGURE 14.22. Tibia vara. A: AP radiograph at age 3 years. B: AP radiograph at age 7 years.
Developmental and Congenital Conditions
 SKELETAL MATURATION
SKELETAL MATURATION
 HERITABLE CONNECTIVE TISSUE DISORDERS
HERITABLE CONNECTIVE TISSUE DISORDERS
Osteogenesis Imperfecta
Other Disorders
 SCLEROSING DYSPLASIAS
SCLEROSING DYSPLASIAS
Osteopetrosis
Osteopoikilosis
Osteopathia Striata
Melorheostosis
 CONGENITAL DWARFISM
CONGENITAL DWARFISM
Achondroplasia
Other Types
 MISCELLANEOUS GENERALIZED CONDITIONS
MISCELLANEOUS GENERALIZED CONDITIONS
Neurofibromatosis
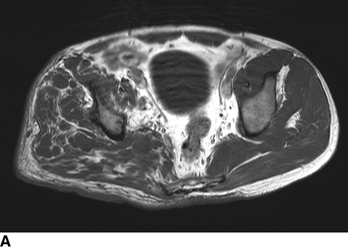
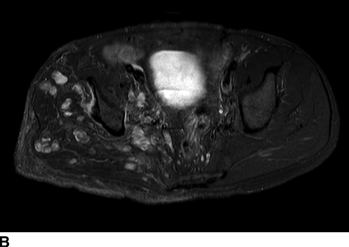
Cerebral Palsy
Muscular Dystrophy
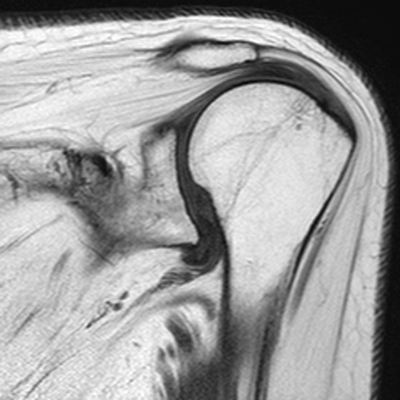
Arthrogryposis Multiplex Congenita
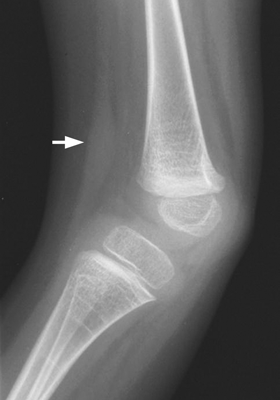
Down Syndrome
Charcot-Marie-Tooth Disease
 MISCELLANEOUS LOCALIZED CONDITIONS
MISCELLANEOUS LOCALIZED CONDITIONS
Multiple Epiphyseal Dysplasia
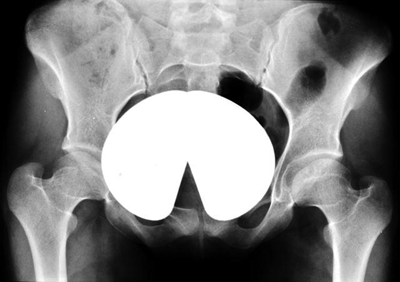
Cleidocranial Dysplasia
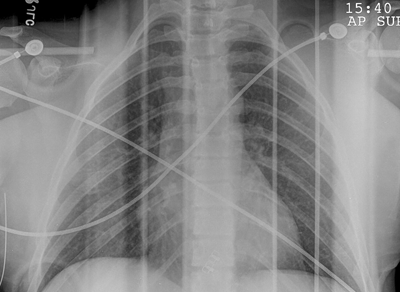
Radioulnar Synostosis
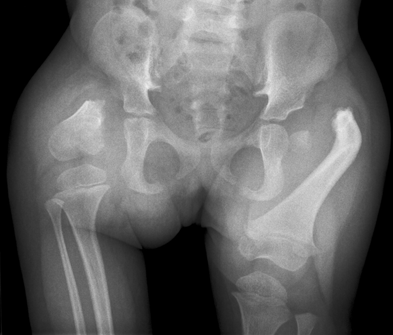
Proximal Focal Femoral Deficiency
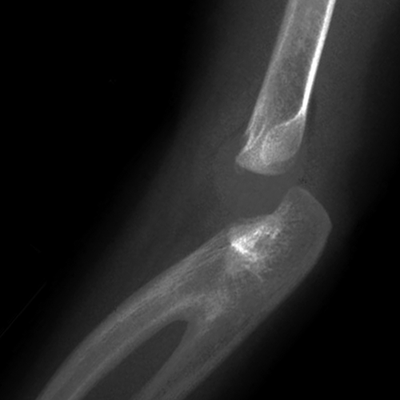
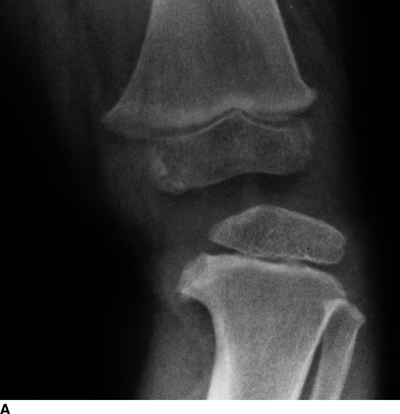
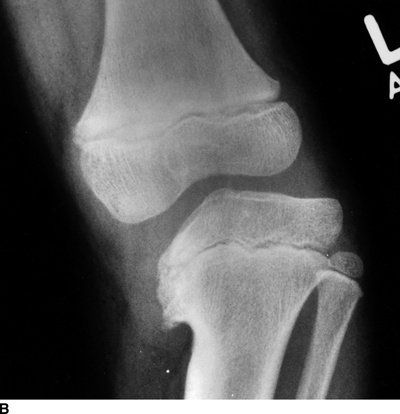
Tibia Vara
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


