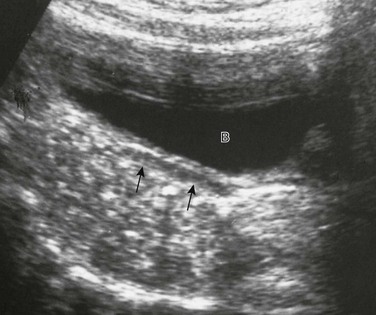
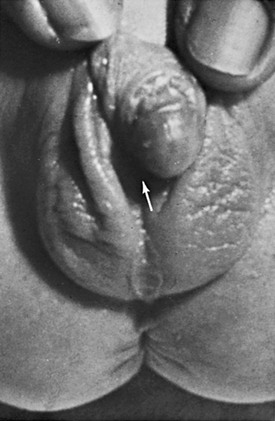
Chapter 125 Overview: The three important precursor components needed for genital system development are the genital ridge and the two sets of internal sex ducts, the müllerian-paramesonephric and the wolffian-mesonephric. Around 7 weeks’ gestation, the embryologic genital ridge becomes either an ovary or a testis. The development of the male genital system is an “active” process requiring testes and müllerian inhibiting substance (MIS). The sex-determining region (SRY gene) on the Y chromosome’s short arm encodes a testis-determining factor. Under this factor’s influence (and in the presence of H-Y antigens found in the cell membranes of normal XY males) there is normal testis development with germ cells in the genital ridge differentiating into Sertoli cells and Leydig cells. Sertoli cells secrete MIS, which causes complete müllerian duct system involution. Leydig cells produce testosterone. The enzyme 5a-reductase converts testosterone intracellularly within the target tissues into the powerful androgen dihydrotestosterone (DHT). DHT allows the wolffian duct system to develop into the epididymis, vas deferens, ejaculatory duct, and seminal vesicles. If no Y chromosome exists or if abnormal encoding of testes determining factor is present, the gonad will passively differentiate into an ovary between 11 weeks to 17 weeks’ gestation in the presence of two X chromosomes. Absence of two X chromosomes may lead to abnormal or streak ovaries. The ovaries and their hormones, however, are thought to have no apparent role in sex differentiation of the female genital tract. The absence of MIS leads to persistence of müllerian structures, which develop into the fallopian tubes, uterus, cervix, and upper vagina. The wolffian ducts involute in the absence of testosterone. The undifferentiated external genitalia include the urogenital tubercle, urogenital swelling, and urogenital folds. DHT stimulation in males causes these structures to develop into the glans penis, scrotum, and penile shaft, respectively. In females, they develop into the clitoris, labia majora, and labia minora, respectively. The prostate gland develops from the urogenital sinus.1–6 Overview: Classic Turner syndrome (isochromatous 45,XO karyotype pattern) is the most common gonadal dysgenesis associated with an abnormal karyotype in girls and is nonfamilial. A single X chromosome is the probable cause for the presence of streak ovaries (streaks or ridges of connective tissue in the mesosalpinges parallel to the fallopian tubes), rather than normal ovaries. Some functional ovarian elements are present in a few cases. Fallopian tubes, a uterus, and a vagina are present, and no wolffian duct derivatives are found.1,5,6 Patients with classic Turner syndrome have several somatic findings. Affected patients are short in stature, with a distinctive facies that includes low-set ears, a low hairline, and a high, arched palate. They have a short, broad, and webbed neck, widely spaced nipples, and a shield chest. Skeletal abnormalities are common—classically short fourth metacarpal, fifth metacarpal, or both. Cystic hygromas of the neck area are noted in fetal life; and the webbed neck is thought to be a residuum. Other variably seen findings include large aortic roots, coarctation of the aorta, renal anomalies (horseshoe kidneys), duplication anomalies, ureteropelvic junction obstruction, and Hashimoto thyroiditis. Patients with Turner syndrome have a history of delayed onset of puberty, no breast development or vaginal mucosal estrogenization (but presence of pubic and axillary hair), infantile internal and external genitalia, and primary amenorrhea.1,7,8 One fourth of patients with 45,XO Turner syndrome have a so-called chromatin-positive pattern. Their karyotype is a mosaic consisting most often of a mixture of 45,XO and 46,XX chromosomes. Less often, other mosaic patterns (e.g., XO/XXX or XO/XX/XXX) or 46,XX and an abnormal X chromosome are present. In such cases, the gonads may consist of a streak ovary on one side and a hypoplastic or normal ovary on the other side, bilateral hypoplastic ovaries, or essentially normal ovaries. External and internal genitalia are entirely female, without wolffian duct remnants. These patients usually do not have the somatic abnormalities typically attributed to classic Turner syndrome, but many are short.1 Patients with mosaic Turner syndrome may develop secondary sex characteristics at puberty (found to occur in about 50%), and some may menstruate regularly.1,9 Primary amenorrhea (defined as a lack of menses by age 16 years), with high levels of circulating follicle-stimulating hormone and luteinizing hormone by serum assays, occurs because of ovarian failure; gonadal tissues fail to respond to endogenous gonadotropins. Patients included in this category have classic Turner syndrome, gonadal dysgenesis (including 46,XY and familial 46,XX), and secondary ovarian failure as a result of radiation or chemotherapy or on an autoimmune basis (autoimmune oophoritis).1,5,6,8 Patients with 46,XY gonadal (X-linked recessive or sex-limited autosomal dominant) dysgenesis are phenotypically female, with streak gonads and infantile internal and external female genitalia. Patients are usually first diagnosed as having an abnormality in adolescence. As with other forms of gonadal dysgenesis, in such patients, the sex chromosome may not be absent but is abnormal. A deletion of the small arm of the Y chromosome (testis-determining factor or MIS) may be present. Mosaicism may lead to the development of ovaries. If a Y chromosome is a component of the karyotype of a patient with gonadal dysgenesis and ovaries, the patient has an increased risk of developing a gonadoblastoma within a dysgenetic ovary. Seminomas also occur in these patients.1,2 Patients with familial 46,XX gonadal (sporadic or autosomal recessive) dysgenesis have gonads that consist of bilateral streaks in some cases, whereas in others hypoplastic ovaries or a hypoplastic ovary may be present on one side and a streak gonad on the other. The internal and external genitalia are entirely female, without wolffian duct derivatives. Incomplete puberty may be observed in patients with residual ovarian tissue. Sexual infantilism and primary amenorrhea are typical findings in those patients with bilateral streak gonads.1,2 Klinefelter Syndrome (47,XXY Seminiferous Tubular Dysgenesis) Seminiferous tubular dysgenesis (Klinefelter syndrome) is the most common aberration of the human sex chromosome. The typical 47,XXY karyotype is found in phenotypic males with primary hypogonadism. It is nonfamilial, occurring in 1 in every 750 to 1000 males. Variants have been described with less common chromosomal abnormalities, including XX/XXY or XY/XXY mosaicism, as well as XXXY, XXXXY, XXYY, or XXXYY sex chromosomal karyotypes. The external genitalia, especially the testes, are small. The testes are usually less than 3 cm in length and are firm. Cryptorchidism and hypospadias are common, with future development of azoospermia and sterility in most patients. The diagnosis is usually not made until after puberty. Gynecomastia develops in almost half of the older patients, and affected patients (particularly those with the classic 47,XXY karyotype) are at an increased risk for breast cancer. Rare cases of testicular and extragonadal germ cell neoplasms have been reported.1,5,6 Persistent müllerian duct syndrome is a rare type of sexual differentiation abnormality in males caused by a deficiency of müllerian inhibiting factor (MIF). These patients usually have a normal 46,XY karyotype and are phenotypically male, but they have a small uterus and fallopian tubes and a small vagina connected to the posterior urethra at the level of the verumontanum. Unilateral or bilateral cryptorchidism is common, as are unilateral or bilateral inguinal hernias. A uterus (uteri hernia syndrome), fallopian tubes, or sometimes a testis may be found in the hernia. The disorder, typically sporadic, has been reported in siblings.1,2,10,11 Pseudohermaphroditism (Intersex) Pseudohermaphroditism or intersex problems are abnormalities in which nonaccord of chromosomal, gonadal, and genital sex is present. Unlike true hermaphroditism, in which two types of gonadal tissue are present, pseudohermaphroditism has nonaccord, but only one gender’s gonads are present. By definition, male intersex patients have testes and female intersex patients have ovaries or ovarian tissue.1 Overview: Female intersex is usually diagnosed in neonatal life in chromosomally normal females (46,XX) with masculinized external genitalia. The cause is usually increased fetal adrenal androgen production, most commonly from congenital adrenal hyperplasia or adrenogenital syndrome. Congenital adrenal hyperplasia (Fig. 125-2) is, by far, the most common cause of abnormal sex differentiation in females, occurring in 1 in 15,000 live births worldwide. It is caused by an inherited deficiency of enzymes involved in adrenocortical hormone biosynthesis. Affected patients have normal ovaries, a uterus, and fallopian tubes and no testicular tissue or internal wolffian duct derivatives. In most cases, the external genitalia are ambiguous (Fig. 125-3), with a prominent penis or partially fused labial scrotal folds. Figure 125-2 Adrenal ultrasound. Imaging: A variously sized vagina is connected with the posterior urethra, forming a urogenital sinus, which commonly empties at the base of the penis. No gonads can be palpated in the labioscrotal folds or in the inguinal canal of such patients because they are located within the pelvis. Voiding cystourethrography (VCUG) often shows a male-type elongated urethra (Fig. 125-4
Disorders of Sex Differentiation
Embryology, Sex Differentiation, and Gonad Differentiation
Disorders of Sex Differentiation Without Ambiguity of External Genitalia (Box 125-1)
Turner Syndrome (XO Gonadal Dysgenesis)
Mosaic Turner Syndrome
Amenorrhea Caused by Hypergonadotropic Hypogonadism
46,XY Gonadal Dysgenesis
Familial 46,XX Gonadal Dysgenesis
Phenotypic Males
Persistent Müllerian Duct Syndrome
Disorders of Sex Differentiation with Ambiguous External Genitalia (Box 125-2)
Female Intersex
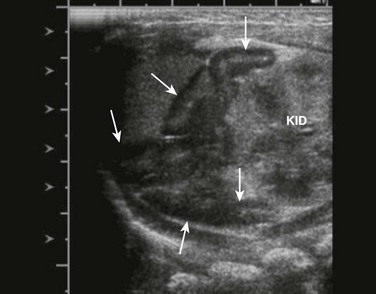
Sagittal view of adrenal and upper left kidney. A very enlarged adrenal (arrows) is seen superior to the upper portion of the kidney. The patient was a newborn with ambiguous genitalia and had three times the normal level of 17 hydroxyprogesterone. Her karyotype proved to be 46,XX. She was diagnosed with salt-wasting 21 hydroxylase deficiency as the cause of her congenital adrenal hyperplasia.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree