Fig. 21.1
Introital cyst. A 10-month-old girl with a persistent vaginal cyst. Intraoperative injection of contrast demonstrated the vaginal cyst (arrow). The cyst was unroofed
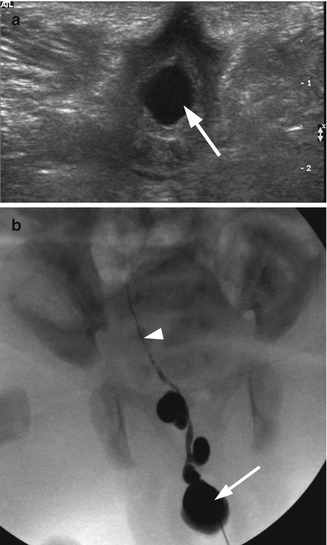
Fig. 21.2
Ectopic ureterocele. A 2-week-old girl with an ectopic ureterocele presented at delivery as an interlabial mass. (a) Transperineal transverse ultrasound demonstrated a cystic mass (arrow) at the introitus. The vagina was not distended. Both kidneys were normal. (b) Intraoperative fluoroscopy with contrast injection to the cystic structure demonstrated an ectopic ureterocele (arrow) and a highly tortuous cystic dysplastic atretic ureter (arrowhead) likely to represent the ureter of an involuted right upper pole
Imperforate Hymen
Imperforate hymen is likely the most common congenital obstructive anomaly in the female reproductive tract. The diagnosis of hydrometrocolpos, or accumulation of fluid in the vagina and uterus, is typically made shortly after birth. The distended hymen presents as a whitish bulge in the posterior introitus and/or a suprapubic mass may be palpable. Whitish vaginal secretions build up in the vagina secondary to maternal estradiol stimulation of the child’s cervical glands. If the diagnosis is delayed after the newborn period, the mucus might be reabsorbed and the bulging hymen no longer evident. If a patient presents in adolescence, it is often with primary amenorrhea, possible cyclic abdominal pain, or a bluish bulging hymen.
Abdominal and pelvic US allows for visualization of the vagina and uterus, as well as the upper urinary tract, which can be obstructed by a dilated vagina (Fig. 21.3). Reimaging is needed to confirm upper tract decompression after an incision of the hymen.
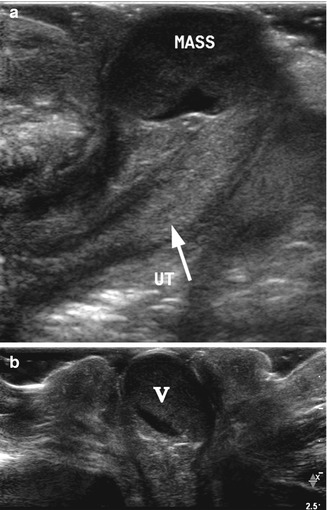
Fig. 21.3
Imperforate hymen. A 1-day-old girl with imperforate hymen presented as a round pink mass protruding through the vagina. (a) High-resolution longitudinal ultrasound demonstrated the uterus (arrow, UT) and a distended vagina appearing as a rounded cystic mass (mass) with echogenic debris. (b) Transverse perineal high-resolution ultrasound demonstrated the distended vagina and the hymen bulging through the introitus
Repair in the newborn consists of a transverse incision of the hymenal tissue. A longitudinal incision would risk injury to the urethra and rectum. Simple aspiration is inadequate as incompletely evacuated material may become infected. In pubertal girls, a hymenectomy of excess tissue may be needed. This condition does not appear to predispose to primary infertility or fetal loss.
Prolapsed Ureterocele
A ureterocele is a cystic dilation of the distal ureter. It is classified as ectopic when it extends distal the bladder neck. If it is large enough, an ectopic ureterocele can prolapse through the urethra, making visualization of the meatus and vagina difficult. With time, the mucosa prolapsing through the urethral meatus changes from pink to congested, dusky purple. In infancy, the ureterocele can prolapse during voiding, resulting in secondary bladder outlet obstruction, urinary retention, and a urological emergency [15]. Management of a prolapsed ureterocele involves manual reduction, often under anesthesia and urethral catheterization for several days. Treatment consists of needle decompression or incision with reduction followed by urethral catheterization. Later, definitive surgical therapy may be required.
About 90 % of ectopic ureteroceles drain the upper pole of a duplicated collecting system. A renal bladder US in the setting of a prolapsed ureterocele is performed to assess for collecting system duplication and upper pole hydronephrosis (Fig. 21.4). Retrograde pyelography can help delineate the relevant anatomy. Subsequent evaluation of the ureterocele also includes a VCUG to rule out concomitant vesicoureteral reflux and bladder abnormalities, as well as a nuclear medicine scan to determine split renal function.
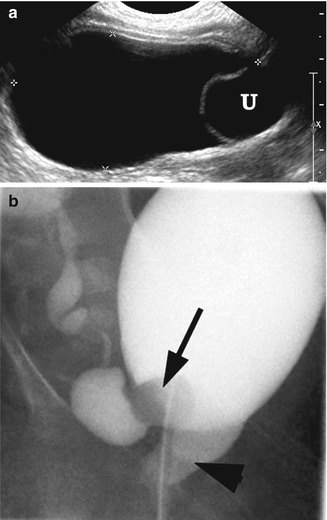
Fig. 21.4
Ectopic ureterocele. A 1-week-old girl with a prolapsed cecoureterocele of right hydronephrotic upper pole kidney. (a) Right longitudinal ultrasound demonstrated distended bladder with ureterocele (U) at the bladder base. (b) VCUG demonstrated a partially everted right ureterocele (arrow) that has prolapsed through the bladder neck to the urethra (arrowhead)
Botryoides Rhabdomyosarcoma Tumor
Rhabdomyosarcoma is the fifth most common malignancy of childhood, accounting for 4–8 % of malignant disease in children under 15 years old [16]. Tumors arise from embryonal mesenchyme, a precursor of striated muscle. Histologic classification includes favorable histology, namely, embryonal rhabdomyosarcoma, with botryoid and spindle cell variants, as well as unfavorable histology, including alveolar, anaplastic, or undifferentiated [17]. About 29 % of cases of rhabdomyosarcoma are genitourinary in origin and most are embryonal [17]. Its botryoid variant has the best prognosis, with a 95 % 5-year survival [17]. Vaginal primary has the best prognosis of all female genital tract primary tumors, owing to favorable histology and early detection [18].
These tumors typically occur in children younger than 4 years old [17]. The classic, and most common, presentation of sarcoma botryoides is an interlabial, prolapsing mass arising from the vagina, bladder, or, rarely, cervix, resembling a “bunch of grapes.” Other presentations include an abdominal mass, bladder outlet obstruction, vaginal discharge, hematuria, bleeding, or passing of tumor fragments [17].
Workup involves a thorough history, physical exam, serum tests, and imaging. Initial imaging is performed with a US to confirm the tumor (Fig. 21.5). Cross-sectional imaging with CT or MRI is recommended to assess the tumor, lymph node involvement, and possible metastases. CT may be superior in assessing abdominal lymphadenopathy, although MRI is often recommended because it lacks ionizing radiation [19]. Limitations of MRI include the following: edema after radiotherapy can be misinterpreted as residual disease in 20 % of cases [20], bright urine on T2 imaging may obscure bladder wall tumors, and, on a delayed post-intravascular contrast scan, gadolinium can confuse interpretation when it layers in the bladder. Therefore, we perform multiphase post-contrast MRI in the evaluation of pelvic masses, which include an arterial, venous, and delayed phase [21]. Importantly, the same modality needs to be used for follow-up after therapy. Contrast cystograms and IVPs are rarely required today. Bone marrow biopsy and bone scintigraphy can assess distant spread. PET is not considered a standard staging modality for rhabdomyosarcoma [22].
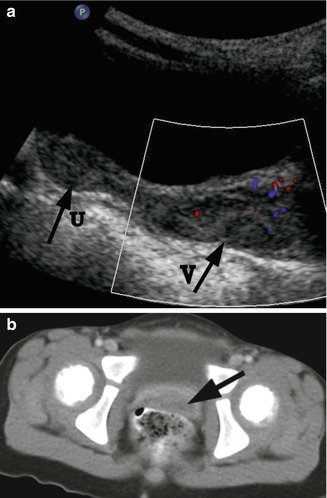
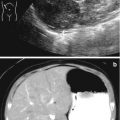
Fig. 21.5
Rhabdomyosarcoma. A 2-year-old girl with a botryoid rhabdomyosarcoma of the vagina presented with a mass protruding from the vagina. (a) US. Longitudinal ultrasound demonstrated a distended vagina (arrow V) with a heterogeneous hypoechogenic solid mass with mild vascularity. The uterus (arrow U) is normal. (b) CT. Post-contrast CT scan demonstrated the vaginal mass (arrow). There was no evidence for lymphadenopathy
Diagnosis requires adequate biopsy, preferably by endoscopy, which is less invasive than percutaneous biopsy and avoids needle tract seeding. If endoscopic biopsy is not possible, percutaneous core biopsy with a Tru-Cut needle or surgical biopsy can also be considered. Fine-needle biopsy is often inadequate. Treatment is multimodal, involving multi-agent chemotherapy, surgical excision, and possible adjuvant radiation. Therapy is aimed at preserving function, especially of the bladder, without affecting survival.
Internal Gynecological Abnormalities
Vaginal Agenesis, or Mayer-Rokitansky-Kuster-Hauser (MRKH) Syndrome
Vaginal, or Müllerian, agenesis is a congenital absence of the proximal vagina in a phenotypically, chromosomally, and hormonally normal female. It is also known as the Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Etiology involves Müllerian aplasia secondary to failure of the sinovaginal bulb to develop and form a vaginal plate. On physical exam, external genitalia are normal. A small vaginal pouch and hymenal fringe are usually present, as they are derived from the urogenital sinus which develops normally. Patients often present with primary amenorrhea, usually without pain.
Vaginal agenesis is associated with variable absence or hypoplasia of the cervix, uterus, and fallopian tubes. An obstructed, normal, or rudimentary uterus with functional endometrium is found in only 10 % of patients [23, 24]. The majority of girls have a solid rudimentary uterus and rudimentary fallopian tubes. Although occasionally cystic, the ovaries are almost always present and functional [25].
Two types of MRKH have been described. In typical (Type A) MRKH, patients have symmetric uterine remnants and normal fallopian tubes, without abnormalities in other body systems. Atypical (Type B) MRKH is characterized with an asymmetric uterus and abnormal fallopian tubes. The atypical form is associated with renal anomalies, such as agenesis or ectopia, and skeletal abnormalities, including scoliosis, cervical vertebrae, and hand and facial abnormalities including scoliosis, cervical vertebral fusion (Klippel-Feil syndrome) as well as hand and fascial abnormalities [26]. The anomalies are part of the Müllerian duct aplasia, renal aplasia, and cervical somite dysplasia (MURCS) association. Unlike cloacal anomalies, vaginal agenesis is not associated with lumbosacral spinal disorders [26].
Surgical management has traditionally included vaginal dimple invagination and skin graft neovagina. Today, vaginal dilation is typically the first line of management, prior to considering surgery. Modern techniques involve vaginal substitution using intestine, made of either sigmoid or ileum [27, 28].
Abnormalities of Vertical Fusion
Transverse vaginal septum is caused by a failure of fusion and/or canalization of the urogenital sinus and Müllerian ducts. Patients present with primary amenorrhea and other symptoms of obstruction, such as abdominal discomfort or pain. Septa are usually less than 1 cm thick and have a small central or eccentric perforation [29]. They most commonly occur in the upper (46 %) and middle (40 %) vagina [30]. Hematometrocolpos from retained menses needs to be treated to improve symptoms and preserve fertility. Patients with a history of transverse vaginal septum have a higher risk of spontaneous abortions, endometriosis, and infertility [31]. Rarely is the etiology of hydrometrocolpos, as seen on US, not obstructive. In our experience, severe vaginal voiding with distension of the vagina with accumulated urine can mimic hydrometrocolpos, with the characteristic resolution once the distended vagina drains by changing body position [32] (Fig. 21.6).
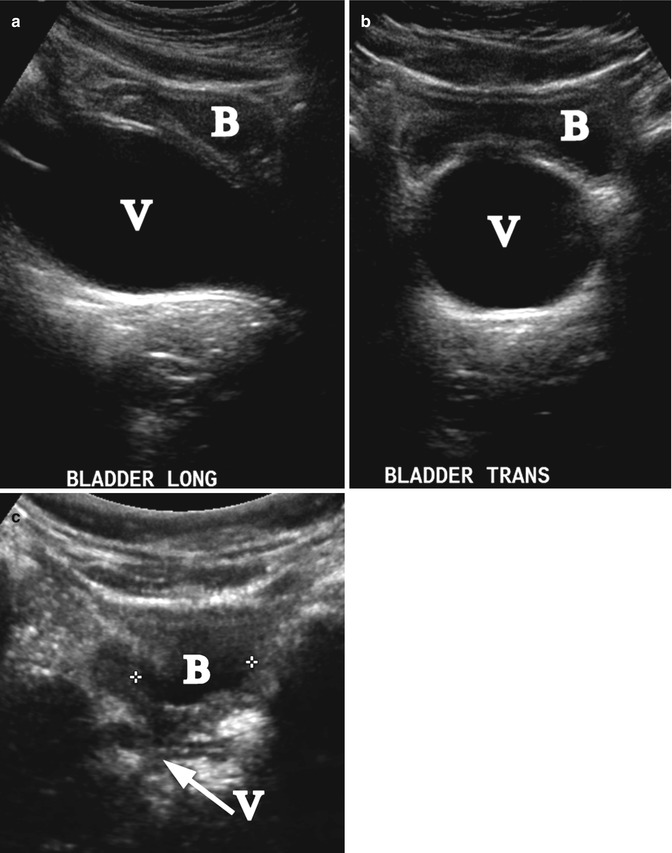
Fig. 21.6
Vesicovaginal reflux. A 6-year-old girl with a UTI and reflux of urine to the vagina mimicking hydrocolpos. Longitudinal (a) and transverse (b) ultrasound demonstrated distension of the vagina (V). The bladder (B) is nearly empty. (c) Repeated post-void ultrasound demonstrated that the vagina (arrow V) was completely emptied (B-bladder)
Transperineal, transrectal, and abdominal US and MRI may help establish the diagnosis and delineate the thickness and location of the septum [9, 10]. In addition, MRI can help determine whether a cervix is present, to differentiate a high septum from a congenital absence of the cervix.
Surgical treatment includes simple incision or excision of the septum with approximation of upper and lower mucosal edges [31] and Z-plasties [33]. Skin grafts to enlarge the vagina carry a higher risk of subsequent vaginal stenosis [34].
Vaginal atresia occurs when the urogenital sinus fails to contribute to the formation of the lower (distal) portion of the vagina. This condition differs from vaginal agenesis or androgen insensitivity syndrome in that the upper vagina, uterus, fallopian tubes, and ovaries are normal. A shallow dimple caudad to the urethral meatus and a distended vagina and uterus may be noted on physical examination. Patients present with primary amenorrhea and pain or a mass secondary to retained menses.
US and/or MRI evaluation is needed to delineate the Müllerian anatomy. Surgical treatment involves a transverse incision at the level of the hymenal ring. After drainage of retained menstrual blood, the vaginal mucosa is identified and a vaginal pull-through performed to bring the distended vagina to the introitus, with possible use of skin flaps and vaginal mobilization.
Abnormalities of Lateral Fusion
Failure of Mullerian duct lateral fusion leads to a spectrum of anomalies of duplicated reproductive structures. True uterine duplication is rare and the two uteri more often present as uterine horns. They range from complete uterine and cervical duplication (didelphys uterus) with or without a longitudinal vaginal septum leading to two vaginas (Figs. 21.7, 21.8, and 21.9), through two uterine horns and one cervix (bicornuate uterus) (Fig. 21.10), to a single uterus with a midline septum and a single cervix (septate uterus).
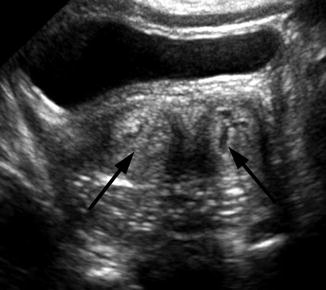
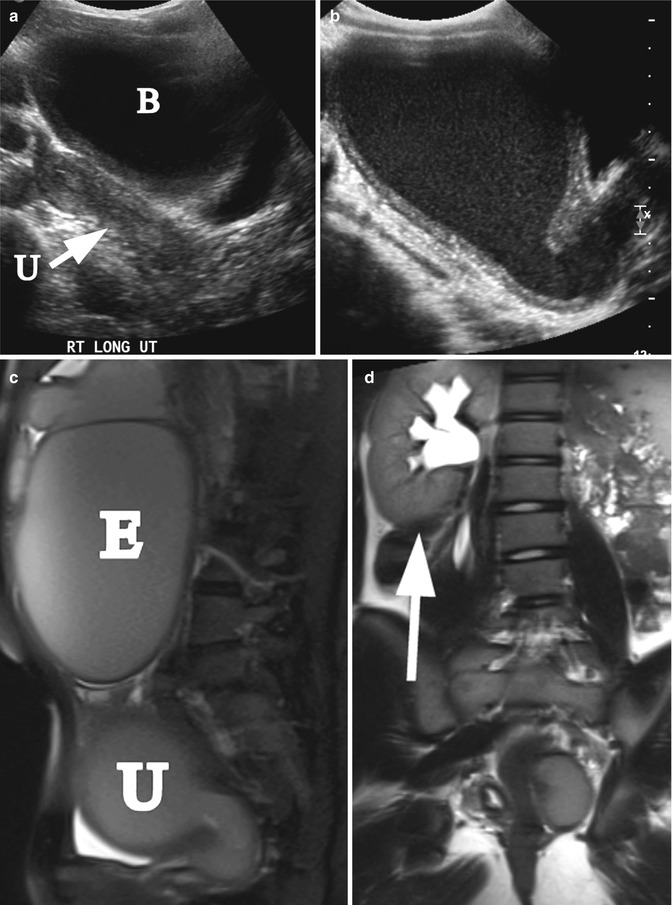
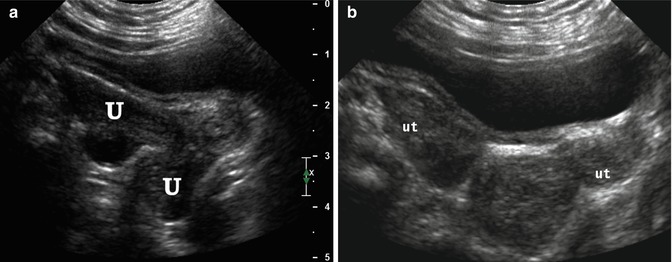
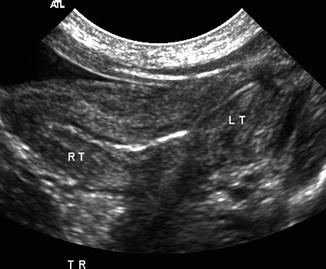
Fig. 21.7
Didelphys uterus. A 1-day-old girl with solitary right kidney and didelphys uterus. Transverse ultrasound demonstrated two separate cervices (arrow)
Fig. 21.8
Didielphys uterus and hydrometrocolpos. A 15-year-old girl with uterus didelphys and left hydrometrocolpos due to distal vaginal agenesis. She presented with abdominal mass and painful menses. (a) Right longitudinal ultrasound demonstrated normal right uterus (arrow, U) and bladder (B). (b) Left longitudinal ultrasound demonstrated hydrometrocolpos. (c) Left sagittal T2 MRI demonstrated left hydrometrocolpos (U) and a large endometrioma cyst (E). (d) Coronal T2 MRI shows a solitary right kidney (arrow)
Fig. 21.9
Didelphys uterus. A 6-month-old girl with didelphys uterus and VACTERL association. Longitudinal (a) and transverse (b) ultrasound demonstrated 2 uteri (U, ut)
Fig. 21.10
Bicornucate uterus. A 1-day-old girl with bicornuate uterus, imperforate anus, cloacal malformation, and solitary right kidney. Transverse ultrasound demonstrated the bicornuate (Rt and Lt, right and left, respectively) uterus
Most patients are asymptomatic, with the exception of uterus didelphys with unilateral vaginal obstruction. Although the patient might experience cyclic or chronic abdominal pain, she does not present with primary amenorrhea because the contralateral vagina is not obstructed. Abdominal US and MRI are excellent modalities to outline these anatomical variants. Renal anomalies, particularly renal agenesis followed by ectopia, are common on the ipsilateral side of the obstructed or maldeveloped system [35] (Fig. 21.8d). Treatment involves a wide transvaginal marsupialization of the vaginal septum to release trapped menstrual blood.
Anomalies in Disorders of Sexual Differentiation
Disorders of sexual differentiation (DSD) require a multidisciplinary approach. A thorough and timely workup allows for appropriate diagnosis, management of any acute medical problems, parental counseling, and gender assignment. It includes the physical exam, imaging, karyotyping, biochemical and hormonal testing, as well as possible laparotomy or laparoscopy with tissue biopsy.
Gonads in individuals with DSD and a Y chromosome are at an elevated risk of developing gonadoblastoma and dysgerminoma. Management involves either a gonadectomy or, with appropriate counseling in children raised male, an orchidopexy. This allows for an early detection of possible malignant transformation (Fig. 21.11).
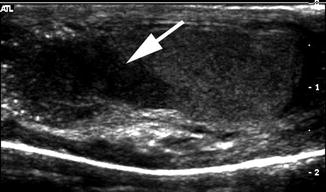
Fig. 21.11
Mixed gonadal dysgenesis with testicular lesion. A 14-year-old child with mixed gonadal dysgenesis and 46XY/45XO mosaicism was raised male and developed intratubular germ cell neoplasm in the right testis. Longitudinal ultrasound of the right testis demonstrated a hypoechogenic mass (arrow)
Complete Androgen Insensitivity Syndrome
Complete androgen insensitivity syndrome (CAIS), formerly known as testicular feminization syndrome, is characterized as a phenotypic female with a 46XY karyotype. Patients present in infancy either with an inguinal hernia or a palpable gonad which turns out to be a testicle or in adolescence with primary amenorrhea. Pubic hair is sparse or absent and the vagina is short and blind-ending without a cervix. Internal female organs are absent. The testes are located along the tract of testicular descent, being intra-abdominal, inguinal or in the labia majora.
The absence of internal female organs can be confirmed with US. Detection of epididymal cysts on an abdominal-pelvic MRI can help in localization and characterization of the gonads (Fig. 21.12). Given a predisposition to gonadal tumors (Fig. 21.13), follow up imaging (US or MRI) is advocated. Other option is surgical exploration offers the benefit of diagnosis with a possible simultaneous prophylactic orchiectomy.
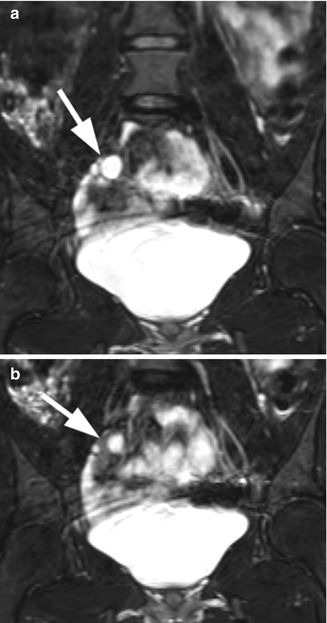
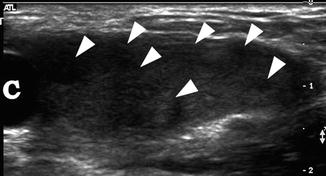
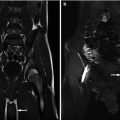
Fig. 21.12
Androgen insensitivity syndrome. An 18-year-old child with androgen insensitivity syndrome and female rearing with multiple epididymal cysts. (a and b) Coronal T2 fat suppression MRI demonstrated the right testis with multiple epididymal cysts (arrows)
Fig. 21.13




Androgen insensitivity syndrome. An 11-year-old child with androgen insensitivity syndrome and female rearing developed Sertoli cell nodular hyperplasia of the left testis. Longitudinal ultrasound of the left groin demonstrated markedly heterogeneous testis with multiple nodules (arrowheads) and an epididymal cyst (c). Epididymal cysts are commonly seen in patients with AIS
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
