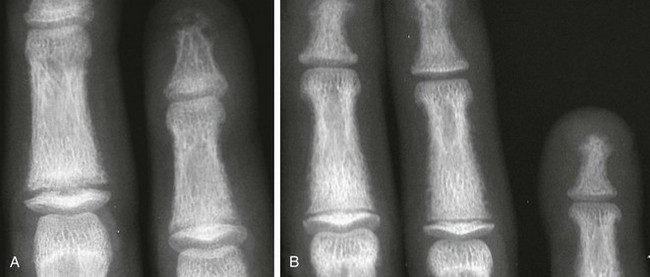
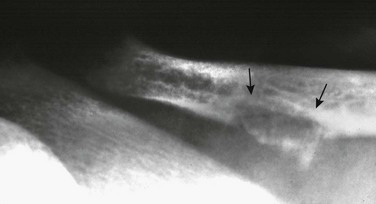
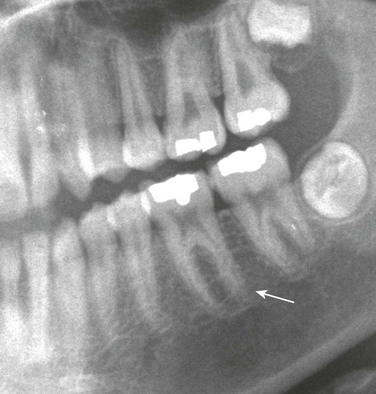
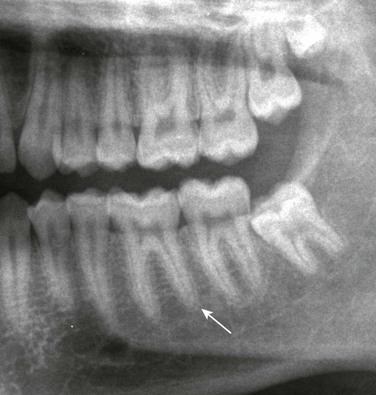
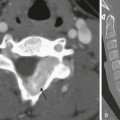
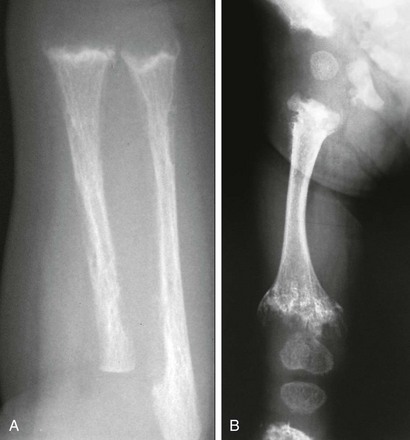
Chapter 141 Several endocrine glands have significant effects on skeletal growth, maturation, and modeling, and understanding these effects facilitates correct radiographic interpretation, allowing for optimal therapy. The parathyroid glands are also intimately involved in mineral homeostasis and have major effects on bone mineralization and resorption.1,2 Because of the importance of hyperparathyroidism (HPTH) in its pathophysiology, renal osteodystrophy is also included in this chapter as are other related conditions such as those causing neonatal hypercalcemia and disorders of bone resorption. Etiology: The parathyroid glands act to maintain a normal circulating calcium concentration. Synthesis and secretion of parathyroid hormone (PTH) are regulated by the parathyroid calcium sensing receptor, which increases PTH in response to hypocalcemia. PTH then has many effects that function to restore the serum calcium concentration. PTH promotes bone resorption to mobilize calcium. In the kidney, PTH upregulates renal 1-α-hydroxylase to produce calcitriol (1,25[OH]2-vitamin D) which then acts on the gut to absorb calcium. PTH also decreases renal calcium excretion and increases phosphate excretion. Renal osteodystrophy (ROD)3–5 comprises a variety of skeletal manifestations of chronic renal failure, the most important of which is secondary HPTH. In chronic renal failure, as the glomerular filtration rate falls, less phosphate is filtered. Phosphate retention and the slight decrease in the ionized calcium concentration caused by it, each stimulates PTH which then acts to promote phosphaturia and increase calcium. Although this initially normalizes calcium and phosphate concentrations, it does so at the expense of elevating PTH and mobilizing calcium and phosphate from bone. With advancing renal failure, decreased renal mass also reduces calcitriol synthesis, leading to rickets and osteomalacia. However, this effect on the skeleton is less pronounced than is HPTH, and many of the manifestations of “renal rickets” are actually those of HPTH. Imaging: PTH stimulation of osteoclasts leads to bone resorption at multiple sites.3–6 The most specific radiographic manifestation of HPTH is subperiosteal resorption, initially seen along the radial aspects of the index and the middle finger middle phalanges (Fig. 141-1). The distal phalangeal terminal tufts are also involved relatively early. With progression, subperiosteal resorption is noted along the ulnar aspects of the phalanges and other bones including the medial aspects of the humeral and femoral necks and the medial aspects of the proximal tibial metaphyses. Although less specific than subperiosteal resorption, HPTH may lead to intracortical resorption, or “tunneling,” causing a striated appearance of the cortex, as well as endosteal, subchondral, and subligamentous resorption (Fig. 141-2). Subphyseal resorption (a form of subchondral resorption) leads to resorption of the zone of provisional calcification and metaphyseal bone beneath the physis, simulating the appearance of rickets. Additional findings of HPTH include resorption of the lamina dura, which forms the thin opaque line surrounding tooth roots (Figs. 141-3 and 141-4), cystic-appearing “brown tumors” (osteoclastomas), and bone sclerosis. Although brown tumors are more characteristic of primary HPTH, overall, they are seen more often in secondary HPTH because it is much more common than primary HPTH in children. Osteosclerosis, most often seen with renal osteodystrophy, may be generalized or most pronounced subjacent to the vertebral end plates, resulting in a “rugger jersey” appearance (Fig. 141-5). Figure 141-5 Rugger jersey spine. Slipped epiphyses (e-Fig. 141-6) are an important complication of renal osteodystrophy in children, accounting for more cases than other predisposing conditions such as hypothyroidism, hypogonadism, and growth hormone deficiency. Although subphyseal resorption can mimic the radiographic appearance of rickets, the radiolucent material beneath the growth plate is fibrous tissue of osteitis fibrosa cystica, which is weaker than the nonmineralized cartilage and osteoid in true rickets, accounting for slipped epiphyses in renal osteodystrophy but not true rickets. Radiographic findings indicating a particularly high risk of slippage include coxa vara with reorientation of the physis from horizontal toward vertical, increased width of the growth plate, and subperiosteal resorption of the adjacent metaphysis. The risk of epiphyseal slippage is particularly high when growth hormone is used to treat short stature, a major clinical problem in children with chronic renal failure. Although slipped epiphyses are seen most frequently in the proximal femur, many other sites may also be involved. e-Figure 141-6 Bilateral slipped capital femoral epiphyses in 11-year-old with renal osteodystrophy. True rickets and osteomalacia may also be seen from decreased production of calcitriol. It is often not possible to distinguish true rickets from the rachitic appearance of osteitis fibrosa cystica. Looser zones, if present, indicate osteomalacia. Renal osteodystrophy usually manifests a “high turnover” state from HPTH. Less often, an adynamic “low turnover” state may also be seen. Previously, adynamic bone disease was often due to aluminum toxicity, which occurred as a complication of dialysis or aluminum-based phosphate binders. Presently, causes of adynamic bone disease include calcitriol therapy (which suppresses PTH), malnutrition, immobilization, corticosteroid therapy, and prior parathyroidectomy. Fractures may occur in renal osteodystrophy with HPTH predisposing to metaphyseal fractures (e-Fig. 141-7). e-Figure 141-7 Metaphyseal fractures in renal osteodystrophy. Treatment and Follow-up: Treatment of secondary HPTH in children with chronic renal disease is directed toward normalization of mineral metabolism to improve growth, decrease skeletal deformity and fragility, and prevent extraskeletal calcification, most importantly vascular calcification.4,7 Treatment is often initiated at stage 3 chronic renal disease (GFR <60 mL/min/1.73 m2) when positive phosphate balance and calcitriol deficiency appear. Dietary phosphate restriction is often difficult to achieve while maintaining adequate protein intake. Hence, intestinal phosphate binders are used. Although calcium-containing phosphate binders such as calcium carbonate have been used extensively, they pose a risk of hypercalcemia and vascular calcification, particular when used along with calcitriol. Aluminum-containing phosphate binders may cause aluminum bone disease and should be avoided. Sevelamer is a newer phosphate binder that does not contain calcium and has been shown to lower phosphate and control the skeletal lesions of HPTH without the adverse effects on vascular calcification that are seen with calcium-containing binders. Primary HPTH in neonates is quite rare and is usually caused by hyperplasia rather than parathyroid adenoma.8 Some cases, neonatal HPTH may result from homozygosity of the gene for familial hypocalciuric hypercalcemia, which usually causes asymptomatic hypercalcemia in adults.9 Additional causes of HPTH in neonates include Jansen metaphyseal chondrodysplasia and I-cell disease. Although Williams syndrome may also cause neonatal hypercalcemia, PTH is not elevated. Jansen metaphyseal chondrodysplasia is an autosomal dominant condition with hypercalcemia and other findings suggestive of severe HPTH during the neonatal period and short-limbed dwarfism later in life. It is caused by a mutation of the receptor for PTH and PTH-related peptide (PTHrP) that causes the receptor to be constantly activated.10,11 Hence, even though no PTH or PTHrP are detectable, PTH signaling is increased, producing effective HPTH with hypercalcemia and bone resorption, which are most prominent early in life (Fig. 141-8). Similarly, constitutive activation of the PTH/PTHrP receptor also leads to excessive PTHrP signaling, which inhibits endochondral ossification by preventing proliferating chondrocytes from entering hypertrophic differentiation.12 This causes an ossification defect leading to buildup of a large amount nonossified cartilage and creating a lucent gap between the epiphysis and the ossified portion of the shaft (see Fig. 141-8). Subsequently, bizarre chondroid calcifications develop within these regions, which become widened and dysplastic. Eventually, the skeleton becomes fully ossified with residual shortening and deformity. The opposite effects are seen in Blomstrand lethal chondrodysplasia with an inactivating mutation of the PTH/PTHrP receptor.13 The lack of PTHrP signaling leads to accelerated endochondral ossification, causing severe growth failure from loss of proliferating chondrocytes and markedly premature physeal closure. Figure 141-8
Endocrine Disorders
Hyperparathyroidism and Renal Osteodystrophy
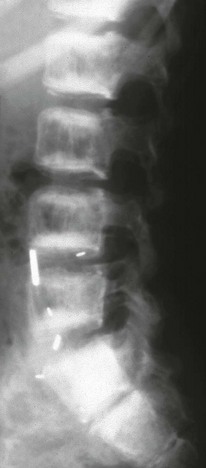
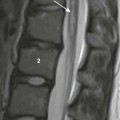
The lateral view of the lumbar spine shows vertebral end plate sclerosis producing rugger jersey appearance.
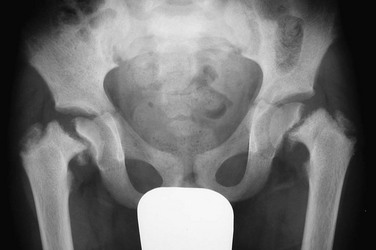
This finding was made at the time of presentation with previously undiagnosed end-stage renal failure.
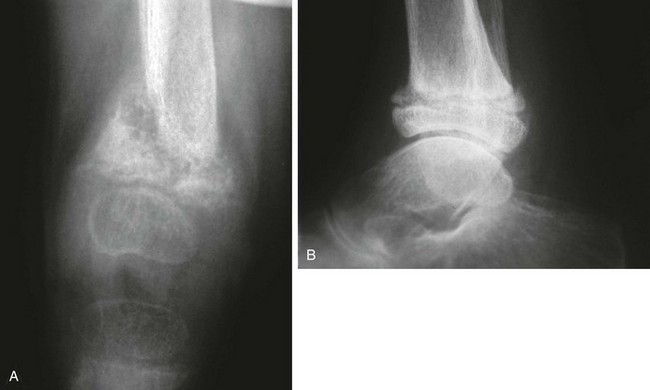
A, Distal femur in a 2.6-year-old child. B, Distal tibial fracture in an 18-year-old.
Other Disorders of Hyperparathyroidism and Causes of Bone Resorption
Jansen Metaphyseal Chondrodysplasia
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Endocrine Disorders