1. Congenital anomalies of the brain suspected or not adequately assessed by sonography including but are not limited to:
(a) Ventriculomegaly
(b) Agenesis of the corpus callosum
(c) Holoprosencephaly
(d) Posterior fossa anomalies
(e) Cerebral cortical malformations
In addition, MRI can be helpful in screening fetuses with a family risk for brain abnormalities such as tuberous sclerosis, corpus callosal dysgenesis, or lissencephaly
2. Vascular abnormalities of the brain suspected or not adequately assessed by sonography including, but are not limited to:
(a) Vascular malformations
(b) Hydranencephaly
(c) Infarctions
(d) Monochorionic twin pregnancy complications
6.2 Technique
Although there are no specific gestational age limits on fetal imaging, it is preferred the fetus be at least 18 weeks both due to the difficulty of imaging such small structures and because of more pronounced motion in younger gestations due to a higher ratio of amniotic fluid to fetus. It is our experience that there is a bimodal demand for fetal MRI, with the first peak before 24 weeks for management decisions and late in the third trimester in preparation for delivery − most often for non-CNS abnormalities. The timing of imaging must be determined on a case-by-case basis per clinical need [2].
All examinations are performed on a 1.5 tesla magnet utilizing a torso coil in most circumstances. Cardiac or body coils can be utilized depending on maternal and fetal size. The mother is placed feet first into the magnet either in prone or left decubitus positioning, per the patient’s comfort. We find the feet first position allows the mother’s head to be closer to the magnet opening, alleviating claustrophobia for some patients. First, 3-plane localizers are obtained relative to the maternal pelvis in the coronal, sagittal, and axial planes. The gravid uterus is then imaged in the maternal axial plane (7-mm slices, 0 gap) with a T2-weighted fast acquisition, typically a half-Fourier single-shot RARE (rapid acquisition with relaxation enhancement) technique, the mainstay of maternal pelvic and fetal imaging because of its high tissue resolution and rapid acquisition times. This ultrafast single-shot technique has an average acquisition time of less than 1 s, depending on imaging parameters. Other names for this sequence include single-shot fast spin echo (SSFSE) and half-Fourier acquisition single-shot turbo spin echo (HASTE), depending on vendor.
In the targeted examination of the CNS, 3-mm slice thickness, 0 gap T2-weighted half-Fourier RARE acquisitions are performed in the coronal, sagittal, and axial planes relative to the fetal brain. The thickness of the slice should be applied based on the size of the fetus, size of the area of interest, and considerations of the trade-off between the potential of improved conspicuity and signal-to-noise ratios. Additional sequences include axial T1-weighted breath hold images and diffusion-weighted images to evaluate for fat/subacute hemorrhage and restricted diffusion/ischemia, respectively. Sequence details can be found in Table 6.2 [23].
Table 6.2
Fetal brain imaging techniques
Acquisition | Plane | Slice thickness (mm) | TR (ms) | TE (ms) | Flip angle | FOV | Matrix | |
|---|---|---|---|---|---|---|---|---|
General | T2 (SS-TSE) | Axial to uterus | 7 | 1100 | 84 | 171 | 300 | 256 |
T1 (SS-SPE) | Axial to uterus | 7 | 128 | 4.76 | 70 | 300 | 256 | |
Fetal brain | T2 (SS-TSE) | Axial, sagittal, coronal | 3 | 1100 | 64 | 150 | 300 | 256 |
T1 (SS-SPE) breath hold | Axial | 5 | 117 | 4.76 | 70 | 300 | 256 | |
DWI | Axial | 5 | 5100 | 104 | 150 | 300 | 192 |
In our practice a radiologist is present at the MRI control panel during all fetal imaging to ensure high-quality images and completeness of the exam. Given the dynamic nature of imaging a moving fetus, the radiologist ensures the images are of optimal orthogonal planes for accurate gestational age measurements and can adjust parameters such as slice thickness or field of view to optimize image quality. The radiologist should also evaluate for extracranial fetal or maternal anomalies that may require additional imaging.
6.3 Fetal CNS Biometry
Biometric measurements including biparietal diameter (BPD) and head circumference (HC) performed by MRI can be used to accurately estimate gestational age with sonographic nomograms [63, 99]. In the evaluation of normal intracranial structures, additional routine measurements include occipital frontal distance (OFD), ventricular atrial diameter, transverse cerebellar width, cisterna magna depth, corpus callosum length, cerebellar vermis height, and inferior vermian distance [62, 99].
The biparietal diameter is utilized to estimate gestational age and is defined on an axial image at the level of the thalami as the distance from the inner table of the skull on one side to the outer table of the skull on the contralateral side, as per ultrasound convention. The occipital frontal distance is also obtained at the level of the thalami and is defined as the midline anteroposterior distance, outer table to outer table (Fig. 6.1). The summation of biparietal diameter and occipitofrontal diameter (the midline anteroposterior distance from outer table to outer table by ultrasound convention) multiplied by π/2 can be used to generate an ellipsoid estimate of fetal head circumference; alternatively the circumference can be obtained directly in some software packages. We utilize the biparietal diameter and head circumference in their respective single variable polynomial equation, designed by Hadlock et al., to fit their respective nomograms for a simple estimate of gestational age [59]. Online calculators are also available. In contrast, obstetric ultrasonographers employ multiple parameters including abdominal circumference and femur length in providing a more reliable estimated gestational age. Significant deviations between an estimated gestational age generated solely by neurologic parameters and the “best” obstetric gestational age by history can indicate micro- or macrocephaly, or intrauterine growth restriction. An estimated gestational age is also crucial to evaluate cortical development (discussed below).
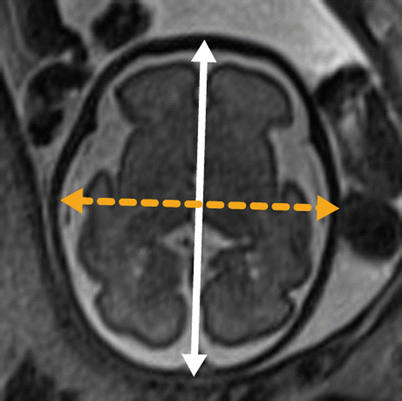
Fig. 6.1
Dashed line represents biparietal diameter (inner table to outer table), and the solid line represents occipitofrontal distance (outer table to outer table)
It is important to note that additional MRI-specific nomograms of the head have been proposed, which define separate cerebral biparietal, calvarial biparietal, and occiptofrontal diameter differently [52]. For the purpose of gestational age assignment, we prefer the more standard inclusion of the skull, as defined by sonographic nomograms. The brain itself can be measured when there is an abnormality and suspected parenchymal underdevelopment and compared to specific nomograms. Care should be taken to ensure the technique chosen matches that of the nomogram utilized.
In addition to the utility in generating an estimate of head circumference, occipitofrontal diameter can be used to calculate cephalic index, a ratio defined by biparietal diameter divided by occipitofrontal diameter, multiplied by 100. A normal cephalic index is between 70 and 85. Cephalic indices larger than this range (brachycephaly) and smaller than this range (dolichocephaly) may indicate the presence of craniosynostosis or syndromic disorder [55] (Fig. 6.2). In a small subset of patients with an abnormal cephalic index, an underlying intracranial abnormality may ultimately be identified.
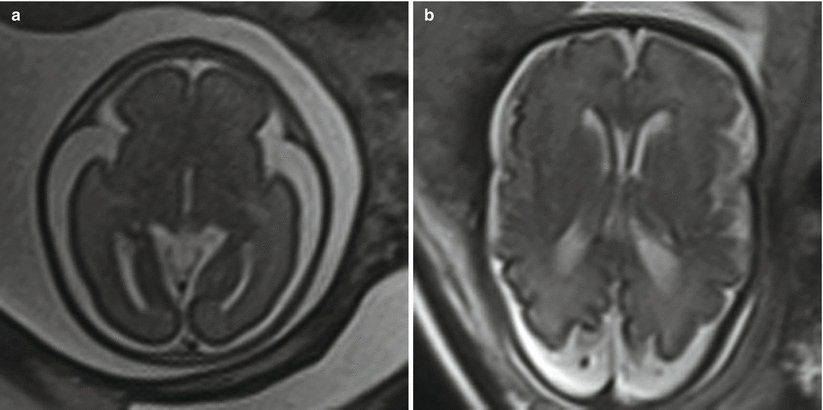
Fig. 6.2
(a) Axial T2-weighted half Fourier images in a 21-week gestational age fetus with a biparietal diameter of 80 mm and an occipital frontal diameter of 87 mm, resulting in cephalic index of 92 %, consistent with brachycephaly. (b) 36-week gestational age fetus with a biparietal diameter of 88 mm, occipital frontal diameter of 132 mm, resulting in a cephalic index of 67 %, consistent with dolichocephaly
Ventricular measurement is typically obtained in the axial plane through the widest transverse segment of the ventricular atria at the level of the thalami, although some prefer measurement in the coronal plane [48, 108] (Fig. 6.3). Transverse cerebellar width, a measurement of greatest transverse cerebellar distance in axial or coronal plane, and vermian length, a measurement of the craniocaudal vermian height in the sagittal plane, aid in identifying underdevelopment of the cerebellum (Fig. 6.4). Assessment of the inferior vermian distance, the anteroposterior distance from the midbrain to the inferior most vermis, is a reproducible method of assessing for vermian elevation (or abnormally increased vermian angle) and should be <5 mm ( Twickler DM unpublished data). Cisterna magna measurements are obtained from the midline posterior aspect of the cerebellar vermis to the inner table of the occiput. Although gestational age dependent, reference values for normal cisterna magna fall between 4 and 10 mm [60]. The corpus callosum should be present by 20 weeks estimated gestational age (Fig. 6.4). Nomograms for multiple components of brain biometry by MRI, including the corpus callosum length and cerebellar vermis, have also been published (Fig. 6.5).
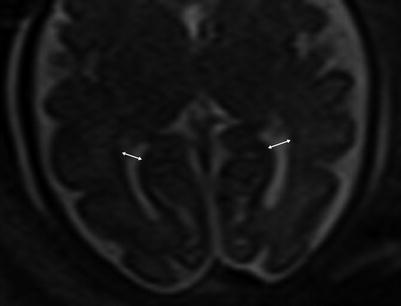
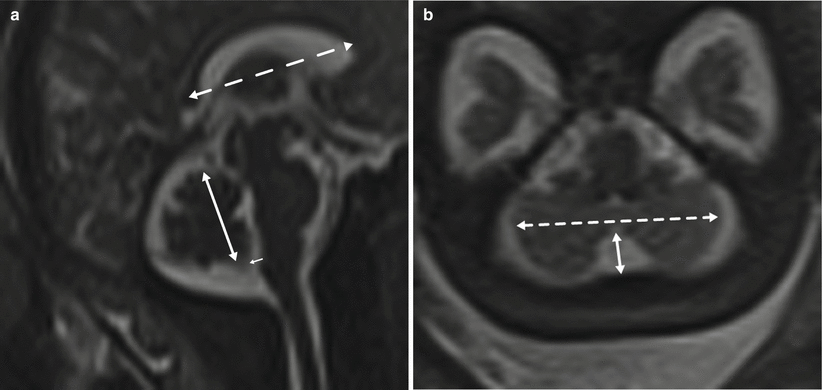
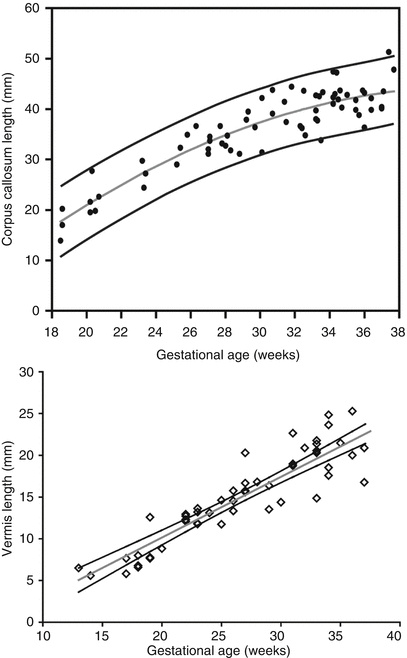
Fig. 6.3
Axial images at the level of the thalami measuring normal lateral ventricles, (Double sided arrow) at 34 weeks gestational age
Fig. 6.4
(a) Sagittal midline image demonstrating the vermian length (solid double arrow), inferior vermian distance (small arrow), and corpus callosum length in a 35-week fetus. (b) Axial image through the posterior fossa in a 34-week fetus demonstrating measurement of the transverse cerebellar distance (dashed arrow) and cisterna magna (solid arrow)
Fig. 6.5
(a) Nomogram depicting corpus callosum length by gestational age (Adapted from Harreld et al. [62]). (b) Nomogram depicting vermian length by gestational age (Twickler DM, unpublished data 2005)
6.4 Cortical Development
Cortical neurons migrate from the germinal zone, the inner most cortical layer, to form the cortical surface. Migration begins around 7 weeks and is mostly complete by 22 weeks [30]. During this span the normal germinal matrix can be seen as a T2 hypointense periventricular band, matching the cortical surface (Fig. 6.6). The developing white matter tissue between the germinal zone and cortical surface, or intermediate zones, demonstrates the opposite signal characteristics, T2 hyperintensity and T1 hypointensity. The germinal zone largely diminishes by 28 weeks [22].
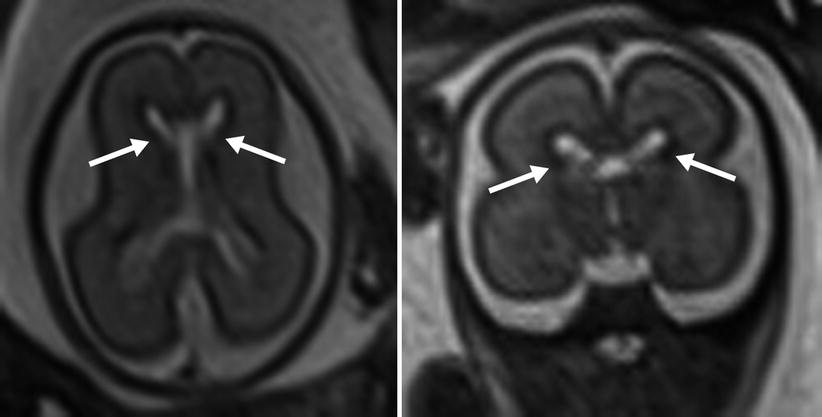
Fig. 6.6
The appearance of the germinal matrix (identified by arrows) at 23 weeks gestational age
Cortical maturation can be evaluated by cerebral gyration and sulcation which tend to occur in a reliable pattern. The MR findings tend to lag behind the actual sulcal development by 1–2 weeks according to pathologic studies [79]. The cerebral hemispheres separate very early in gestation, a process that is typically complete by 12 weeks. The sylvian fissure and lateral sulcus are next to develop and are typically present by 20 weeks gestational age. The parietooccipital fissure is also usually present by 20 weeks. Beyond 20 weeks sulcation and gyration accelerate. The formation of the calcarine fissure is consistently visible around 24 weeks, the central sulcus usually by 26 weeks, precentral by 27 weeks and the postcentral by 28 weeks. Sulcation of the frontal and temporal gyri is usually well underway by 32 weeks and is required for normal development of the sylvian fissures. Some of the major sulcal milestones can be found in Fig. 6.7 and in our opinion are most easily assessed on sagittal and parasagittal images, although all planes can be useful. Twin pregnancies can result in delayed gyration of 2–3 weeks [29].
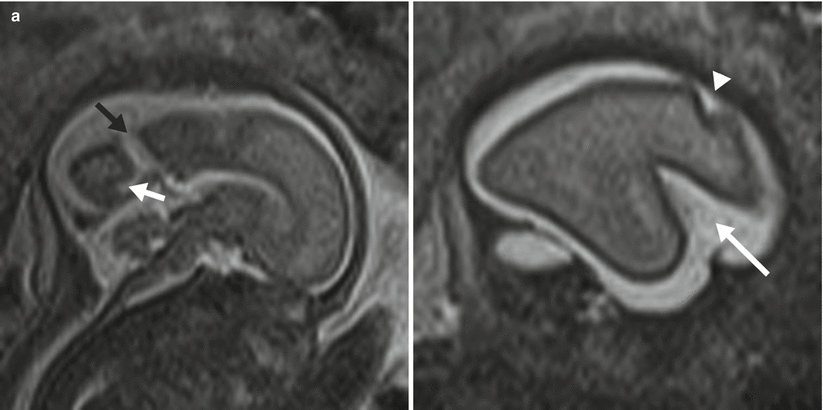
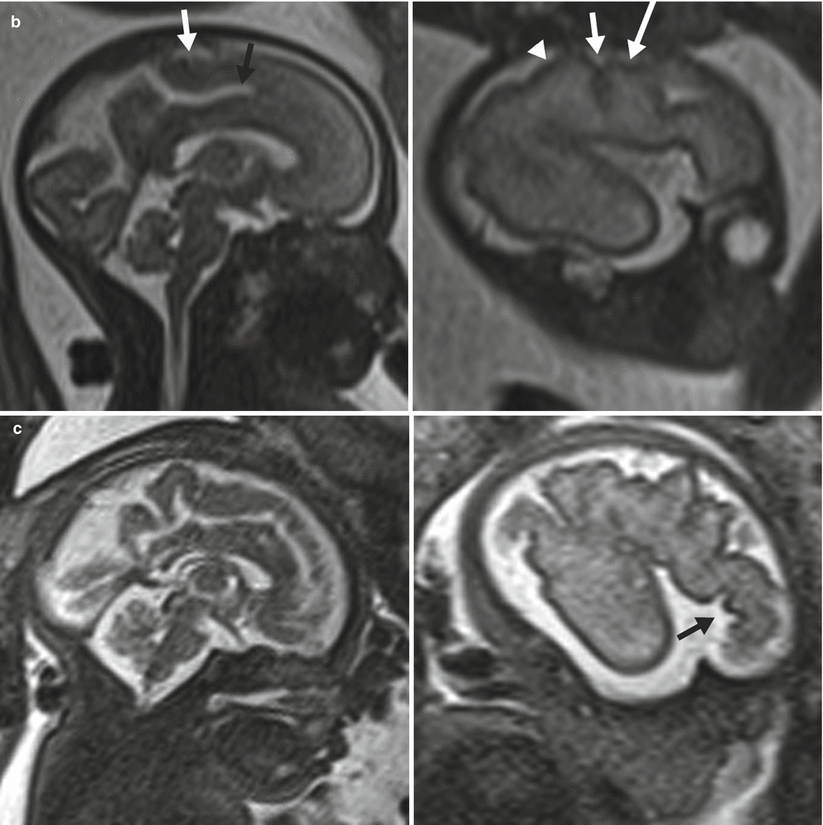
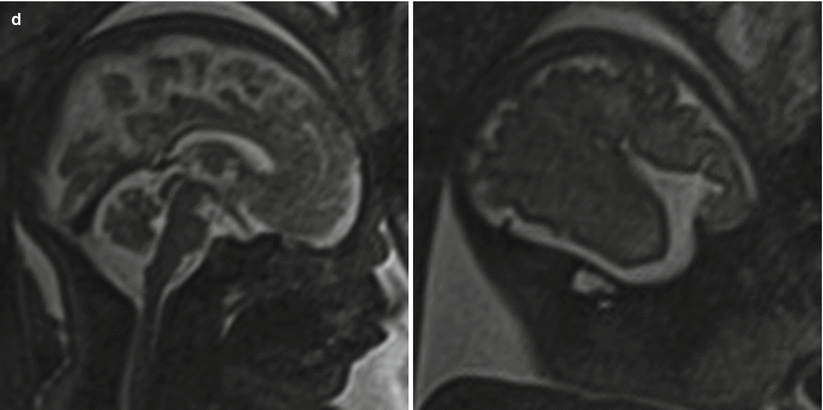
Fig. 6.7
(a) A 26-week fetus demonstrating parietooccipital sulcus (black arrow), calcarine sulcus (white arrow), central sulcus (white arrowhead), lateral sulcus, and sylvian fissure (long white arrow). (b) 28-week fetus demonstrating central sulcus (white arrow), cingulate sulcus (black arrow), precentral sulcus (long white arrow), and postcentral sulcus (white arrowhead). (c) A 32-week fetus and (d) a 34-week fetus demonstrating progressive interval gyration and sulcation. The black arrow in (c) identifies the developing frontal operculum
Myelination is the final stage of cerebral development, beginning during the second trimester and proceeding into the third decade of life [19]. Diffusion anisotropy has the potential to characterize pre-myelinating structures and can identify the corpus callosum and other white matter tracts [23]. At this point in time, more work needs be done to evaluate clinical utility in the developing fetus.
6.5 CNS Pathologies
6.5.1 Ventriculomegaly
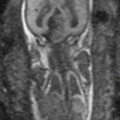
Ventriculomegaly is one of the most common cerebral anomalies in the fetus and likely the most frequent indication for fetal MRI. The term ventriculomegaly describes the finding of lateral ventricular enlargement and is typically more accurate at the point of patient referral than the term hydrocephalus, a diagnostic term describing enlarged ventricles secondary to elevated ventricular pressures. A broad array of diagnoses can result in the imaging appearance of ventriculomegaly, and MRI aids in identifying the etiology and additional structural abnormalities, allowing for more precise patient counseling [107]. Ventriculomegaly is defined by enlargement of one or both ventricular atria to >10 mm (4 standard deviations above the mean) and is subdivided into categories of mild (10–12 mm), moderate (13–15 mm), and severe (>15 mm) [35, 45, 102, 104] (Fig. 6.8).
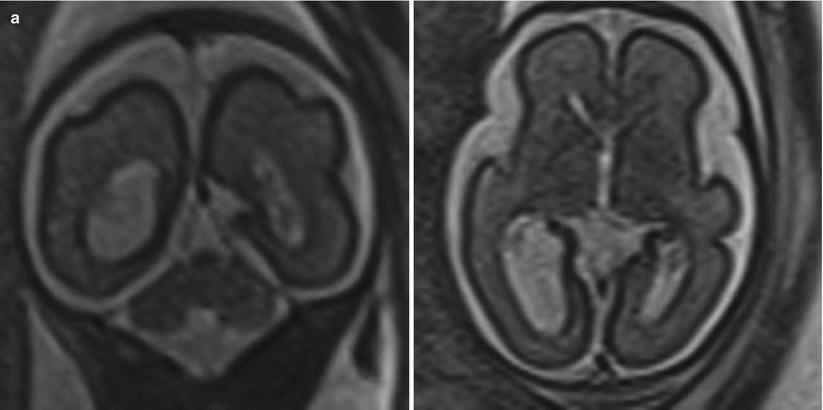
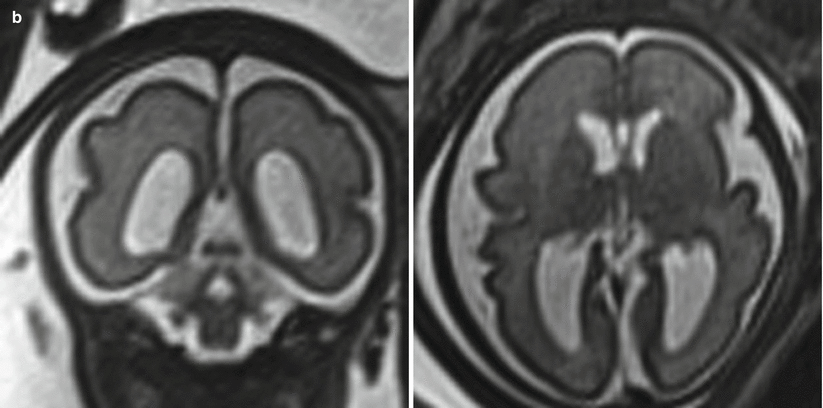
Fig. 6.8
(a) Mild isolated right ventriculomegaly of 12 mm in a 25-week fetus. (b) Moderate bilateral ventriculomegaly measuring 13 mm bilateral in a 29-week fetus with confirmed trisomy 21
The etiologies of fetal ventriculomegaly seen with imaging can be broadly classified into obstructive causes, neurologic malformations, and secondary to destructive processes. Effacement of subarachnoid spaces in the setting of ventriculomegaly is suggestive of obstructive hydrocephalus. Aqueductal stenosis is an obstructive cause of ventriculomegaly and can be recognized by distention of the lateral and third ventricles with a normal-appearing fourth ventricle and posterior fossa. Ventricular distention in aqueductal stenosis typically progresses with gestational age. There are multiple etiologies of aqueductal stenosis, including congenital, associated X-linked or autosomal recessive disorders, hemorrhage, and infection. X-linked aqueductal stenosis is the result of a mutation of Xq28, a gene responsible for a neural cell adhesion molecule [43]. X-linked aqueductal stenosis is nearly always associated with severe mental retardation and can be suggested by imaging in the setting of a male fetus with adducted thumbs [61] (Fig. 6.9). Other obstructive processes include Chiari 2 malformation, intraventricular hemorrhage, and mass effect from cystic or solid cerebral masses. Ventriculomegaly secondary to cerebrospinal fluid overproduction can be seen in the setting of choroid plexus papillomas, benign tumors which can have severe neurologic consequences secondary to their large size, and cerebrospinal fluid hypersecretion [25].
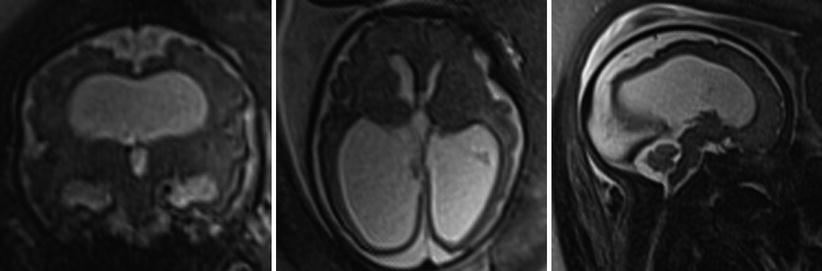
Fig. 6.9
Images of a 31-week male fetus with severe ventriculomegaly but normal-sized fourth ventricle in this patient with X-linked aqueductal stenosis
Additional cerebral anomalies can be found in 50–60 % of fetuses with ventriculomegaly and are more likely in proportion to severity of ventriculomegaly [45, 56, 75, 116]. The most common associated anomalies are Chiari 2 malformation, agenesis of the corpus callosum, Dandy-Walker malformation, holoprosencephaly, mass lesions, and destructive processes [31, 46]. Non-CNS abnormalities are present in one-third of cases [45]. Associated abnormalities are more common with more severe ventriculomegaly and are also associated with a worse prognosis [54, 117].
Isolated ventriculomegaly, cases of ventriculomegaly without additional abnormality, is the most common ultimate diagnosis [92]. Prognosis is dependent on the severity of ventriculomegaly. Normal neurologic outcomes are seen in 93–96 % of mild ventriculomegaly, 76 % of moderate ventriculomegaly, and 28 % of severe ventriculomegaly [46, 104, 116]. The incidence of perinatal and neonatal death also correlates with the degree of ventriculomegaly [45]. Ventricular enlargement throughout pregnancy is associated with increased incidence of developmental delay. Outcomes in asymmetric ventriculomegaly are also dependent on the severity of distention of the affected ventricle and can be equally good or poor when compared to symmetric ventriculomegaly [90, 116]. The risk of aneuploidy is low in the setting of isolated mild ventriculomegaly (1.8 %) [90], and thus far, the severity of ventriculomegaly has not been linked to increased risk of aneuploidy [45]. Intrauterine infection also appears to be an infrequent cause of ventriculomegaly (1.8 %) [90, 116]. Additional imaging findings related to CNS infection will be discussed later in this chapter.
A few authors have found male fetuses to have larger atria than female fetuses on average and a lower prevalence of developmental delay [93, 95, 116]. Other authors have found no significant differences between the sexes, suggesting this data be used with caution when counseling [58, 78].
It should also be noted that extreme ventricular dilatation can result in a thinned corpus callosum. Careful inspection for the presence or absence of the corpus callosum should be made in these cases as absence of the corpus callosum conveys its own associated anomalies.
Malformations of the developing brain can be divided into three broad categories: abnormal neurogenesis, abnormal neuron migration, and abnormal neuronal organization.
6.6 Abnormal Neuronal Migration
6.6.1 Callosal Dysgenesis and Agenesis
Fetal MR provides for the direct visualization of the entirety of the corpus callosum, the largest of the cerebral commissures. Normal development of the corpus callosum is a sensitive indicator for normal brain development. In studies comparing MRI to ultrasound for the evaluation of callosal abnormalities, MRI detects additional CNS abnormalities in up to 22.5 % of cases (most commonly posterior fossa malformations and cortical malformations) [105]. The corpus callosum begins forming around the 11th week of gestation and is typically completed by the 21–22th week [1, 97]. The corpus callosum is composed of five named regions: rostrum, genu, body, isthmus, and splenium. Malformation (hyperplasia, hypoplasia, or agenesis) of any region is termed callosal dysgenesis (Fig. 6.10); complete absence is termed agenesis of the corpus callosum.
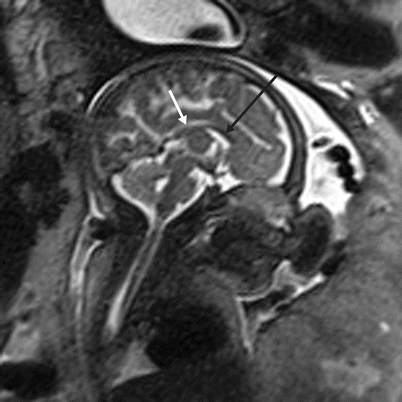
Fig. 6.10
Callosal dysgenesis. Sagittal T2-weighted half Fourier RARE of a 31-week male fetus with thinned genu (black arrow) and body (white arrow) of the corpus callosum. The splenium and rostrum are not seen
The causes of callosal dysgenesis and agenesis are numerous and heterogeneous, as commissural formation relies on a complex series of steps that are each prone to insult/error. Complete agenesis is estimated to have a prevalence of 0.01–0.3 % [53, 114] and be found in 2–3 % of individuals with developmental disorders [70].
In complete agenesis both axial and coronal images will fail to depict crossing callosal fibers. The lateral ventricles will appear unusually parallel on axial images and often meet the criteria for moderate or severe ventriculomegaly. Unless ventriculomegaly is progressively worsening throughout pregnancy, concomitant ventricular enlargement does not portend a worse prognosis [51]. The cavum septi pellucidi, a space between thin walls of tissue that connect between the fornices and a normally formed corpus callosum, will not be seen. Coronal images demonstrate teardrop-shaped lateral ventricles (termed colpocephaly). Enlargement and rostral displacement of the third ventricle may appear to communicate to the median interhemispheric fissure. Midsagittal images will depict sulci radiating outward from the expected location of the corpus callosum (Fig. 6.11). Midline interhemispheric cysts are sometimes seen in the expected location of the cavum septi pellucidi and may or may not be in communication with the ventricular system (termed Types 1 and 2, respectively) [16]. If enlarged, these cysts can result in hydrocephalus or macrocephaly. Additional concomitant findings include periventricular nodular heterotopia, pericallosal lipomas, vermian hypoplasia, porencephaly, mega cisterna magna, nodular heterotopia, and abnormalities of sulcation and neuronal migration [76, 106].
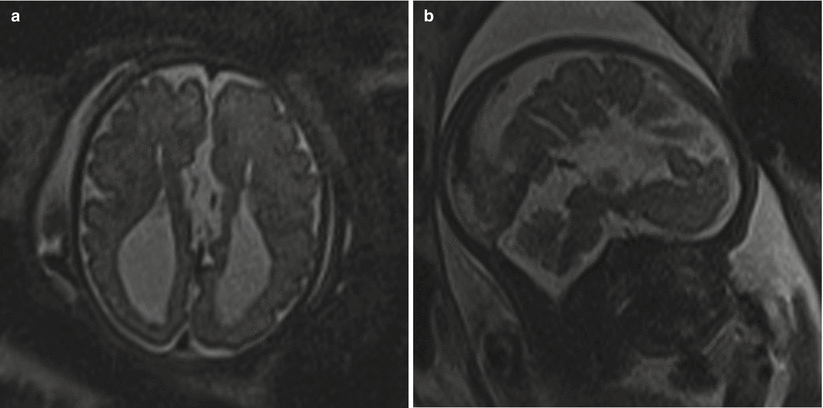
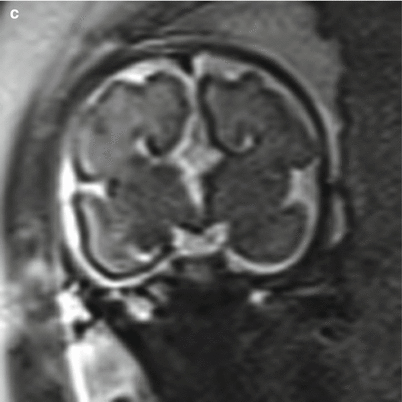
Fig. 6.11
(a) T2 half Fourier RARE images of a 32-week fetus with an axial image demonstrating parallel lateral ventricles and colpocephaly, technically consistent with moderate bilateral ventriculomegaly. (b) Sagittal image through the midline depicting the radiating ray appearance. (c) Coronal T2 half Fourier RARE images obtained in a different 31 weeks estimated gestational age demonstrating lack of crossing callosal fibers and the viking helmet or longhorn appearance of the lateral ventricles
Callosal dysgenesis and agenesis are associated with numerous developmental malformations and syndromes, with the prognosis dependent on what additional findings are present. In a large California-based population study of fetal MR findings of callosal dysgenesis and agenesis, chromosomal anomalies were seen in 17.3 %, with 49 % having additional CNS abnormalities including cortical malformations, posterior fossa abnormalities, and intracranial cysts [53, 106]. When MRI, genetic, and serologic workup is negative for additional anomaly, callosal agenesis is described as isolated. Neurodevelopmental outcomes are normal in approximately 75 % of fetuses with isolated callosal agenesis, with the remaining 25 % at increased risk for moderate to severe learning disabilities [51, 105].
6.6.2 Lissencephaly Spectrum
Lissencephaly is a disorder of neuronal production or migration resulting in absence of gyral formation (agyria) or reduced gyration (pachygyria). Because the brain is agyric before 22 weeks, these malformations are often difficult to identify as significantly delayed before 30 weeks. Classic lissencephaly can be both inherited and sporadic, associated with isolated gene defects and Miller-Dieker syndrome – an autosomal dominant defect on chromosome 17p resulting in characteristic dysmorphic facies (wrinkled skin over the glabella, narrow forehead, prominent occiput, and small chin), severe mental retardation, and death in infancy or early childhood (OMIM #247200). In addition to a smooth gyral pattern, ventriculomegaly or colpocephaly and callosal dysgenesis agenesis are also seen in classic lissencephaly [32, 40]. The term variant lissencephaly describes a set of disorders morphologically less severe in appearance as compared to classic lissencephaly but with similarly poor neurologic outcomes [41]. The cobblestone complex (formerly termed Type 2 lissencephaly) refers to a disorganized “cobblestone” cortex and in contradistinction to classic lissencephalies is due to excessive neural migration secondary to defective glycosylation. These syndromes demonstrate a variable degree of pachygyria, gliotic white matter, and cerebellar hypoplasia. Associated diseases include the autosomal recessive Walker-Warburg syndrome, muscle-eye-brain disease, and Fukuyama congenital muscular dystrophy [71, 109].
6.7 Abnormal Postmigrational Organization
6.7.1 Polymicrogyria
Polymicrogyria is the result of abnormal post-migrational organization into numerous gyri and can result from infection (particularly CMV), maternal hypotension, and inherited disorders [10, 27]. Polymicrogyria appears as focal or diffuse abnormal infoldings of the T2 hypointense cortical surface and most commonly involves the frontal lobes and sylvian fissures. Polymicrogyria is also more commonly bilateral (Fig. 6.12). Polymicrogyria is associated with developmental delay, epilepsy, and focal neurologic defects depending on the involved structures [7, 64].
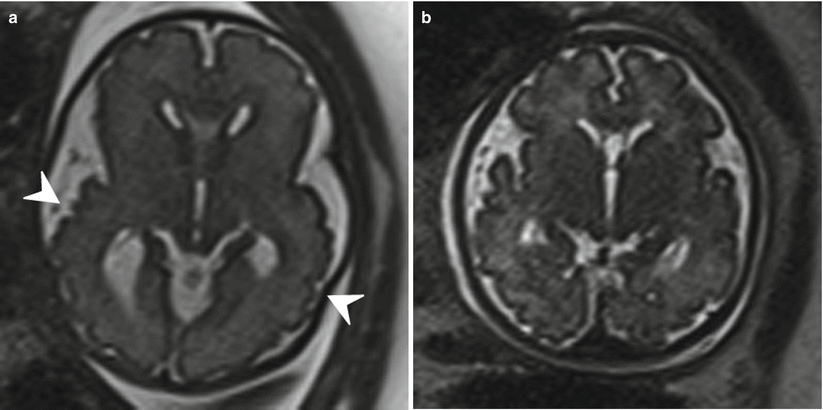
Fig. 6.12
(a) Axial T2 half Fourier RARE images obtained at 35 weeks estimated gestational age depicting bilateral polymicrogyria (arrowheads). (b) Normal axial T2 half Fourier RARE image for comparison
6.7.2 Schizencephaly
Schizencephaly is the result of abnormal neuronal migration and results in parenchymal clefts lined by dysplastic cortex that communicate with the lateral ventricles and can be unilateral or bilateral. Mutations altering the expression of the EMX2 homeobox gene in proliferating neuroblasts have been associated with schizencephaly [24]. Infectious, traumatic, and toxic etiologies are also suggested [89]. In Type 1, or closed-lip schizencephaly, the walls of the cleft are nearly apposed to each other with the parenchymal cleft communicating with the pia-ependymal seam, a structure continuous with the lateral ventricle. In Type 2, or open-lip schizencephaly, large cleft extends from the cortical surface to the lateral ventricle (Fig. 6.13). The identification of gray matter lining the clefts differentiates schizencephaly from large areas of parenchymal destruction seen in porencephaly. Additional findings sometimes seen include absent cavum septum pellucidum and ventriculomegaly. Polymicrogyria and optic hypoplasia are also associated but are usually identified postnatally. The degree of impairment depends on the size, bilaterality of the lesions, and the involved structures, with large bilateral defects having a worse prognosis. Clinical sequelae can include developmental delay, paralysis, and seizures. Seizure disorder but otherwise normal neurodevelopmental outcome has been seen in patients with small closed-lip defects [11]. As many as 47 % of patients with the appearance of an open-lip defect will have a closed-lip defect postnatally [87].
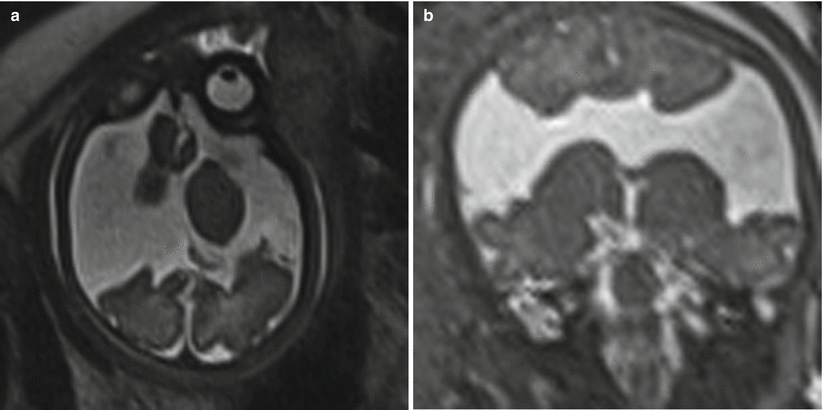
Fig. 6.13
Axial (a) and coronal (b) T2-weighted half Fourier RARE images demonstrating bilateral open-lip schizencephaly in a 37-week gestational age fetus
6.7.3 Subependymal Lesions
Gray matter heterotopia describes the abnormal localization of gray matter as a result of failure of neuronal migration, resulting in periventricular (subependymal), subcortical, and leptomeningeal heterotopia. Periventricular heterotopias occur by sporadic or multiple inherited genetic defects [36]. At imaging nodules of tissue isointense to the cortex outline the ventricles, projecting into the ventricular lumen, and may be focal or diffuse (seen in the autosomal recessive form Xq28 gene defect) (Fig. 6.14) [84]. Care must be taken to not confuse an involuting germinal matrix, which occurs between 26 and 28 weeks gestation, and will not protrude into the lateral ventricles. Subependymal hamartomas of tuberous sclerosis are a differential consideration and appear hyperintense on T1-weighted images [17]. There may be mild to moderate ventriculomegaly and additional findings of callosal agenesis and rarely encephaloceles. Subcortical and leptomeningeal heterotopias also occur, but have rarely been documented antenatally due to the limited spatial resolution of MRI.
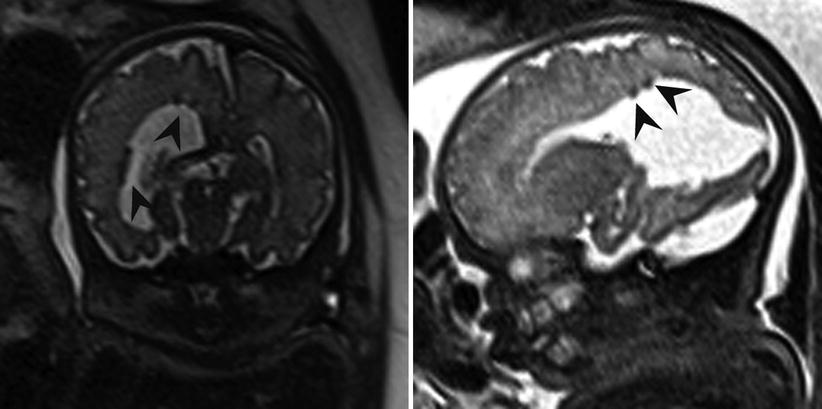
Fig. 6.14
Axial and coronal T2-weighted half Fourier RARE images demonstrating subependymal heterotopia (arrowheads) in a 34-week fetus with right posterior open-lip schizencephaly
6.7.4 Abnormal Neurogenesis
Microcephaly is a descriptive term rather than a diagnosis and may be due to a primary disorder of neurogenesis or secondary to systemic insult or injury. Microcephaly is defined by most as a head circumference less than 3 standard deviations below the mean for gestational age (Fig. 6.15). In the primary forms of microcephaly (microcephaly vera), additional findings often seen include a simplified gyral pattern (microlissencephaly), callosal dysgenesis, and gray matter heterotopia [73]. Secondary microcephaly can be the result of ischemic events, phenylketonuria, fetal alcohol syndrome, infections (CMV, toxoplasmosis), and additional toxins.
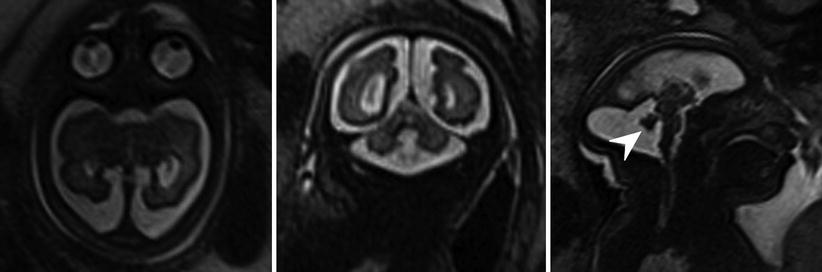
Fig. 6.15
Axial, sagittal, and coronal T2-weighted half Fourier RARE images microcephaly with microlissencephaly in a 35-week gestational age fetus. Anterior sloping of the forehead and vermian hypoplasia (arrowhead) are also seen in this case
Megalencephaly describes increased brain volume and to our knowledge has not been diagnosed antenatally. The few etiologies of megalencephaly described have been collectively categorized as megalencephaly capillary malformation-polymicrogyria syndromes until more is discovered about their pathogenesis [9]. Hemimegalencephaly is hamartomatous overgrowth of a cerebral hemisphere and is distinct from the etiologies of megalencephaly [38]. Hemimegalencephaly is sporadic and also associated with a number of syndromes including Klippel-Trenaunay-Weber, epidermal nevus, and Proteus syndrome. Children typically demonstrate severe mental retardation, seizures, and hemiplegia [37].
6.8 Posterior Fossa Malformations
6.8.1 Fluid Collections of the Posterior Fossa
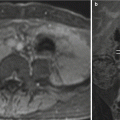
A common indication for fetal MRI is cystic expansion of the posterior fossa, an ultrasonographic finding that elicits an evaluable differential diagnosis list with different clinical implications and commonly includes Dandy-Walker malformation, hypoplastic cerebellar vermis, Blake pouch cyst, mega cisterna magna, and arachnoid cysts. Depending on the extent of the cystic expansion and associated mass effect, distinguishing between these abnormalities can be challenging and may not be possible if cyst walls are too small for visualization.
A Dandy-Walker malformation is classically described as a triad of partial or complete vermian agenesis, an enlarged posterior fossa with elevation of the tentorium and confluence of sinuses (e.g., torcula), and a large cystic appearing in the fourth ventricle which extends dorsally (Fig. 6.16). Mass effect from the cyst often results in compression of the brainstem against the clivus. Dandy-Walker malformations are associated with de novo deletions of 3q and have not demonstrated Mendelian inheritance patterns [57, 86]. The term Dandy-Walker variant, historically utilized to describe vermian hypoplasia without enlargement of the posterior fossa, has fallen out of favor as more recent embryologic data suggests each of the findings in Dandy-Walker malformation can have different degrees of severity but are attributable to the same genetic aberrations [88]. The term Dandy-Walker complex is preferred for when a similar constellation of findings are present, but to a lesser severity than the findings of true Dandy-Walker malformation. Commonly associated cerebral abnormalities include ventriculomegaly, callosal dysgenesis, occipital encephaloceles, and gray matter heterotopias. Dandy-Walker malformation in the presence of additional developmental defects is often associated with chromosomal abnormalities (up to 46 %) [34, 45]. Neurodevelopmental outcome in children with isolated Dandy-Walker malformation is variable and has most commonly been associated with mental retardation, hypotonia, and epilepsy. In milder expressions of Dandy-Walker complex, outcomes may be normal in 50–80 % of patients , with normal vermian lobulations [47, 77].
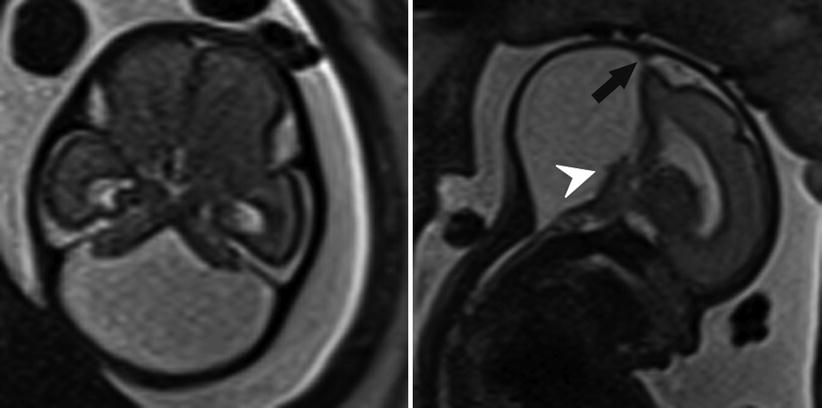
Fig. 6.16
Axial and sagittal T2-weighted half Fourier RARE images of a 26-week gestational age fetus with severe vermian hypoplasia (white arrowhead), an elevated torcula (black arrow), and cystic dilatation of the posterior fossa, consistent with Dandy-Walker malformation
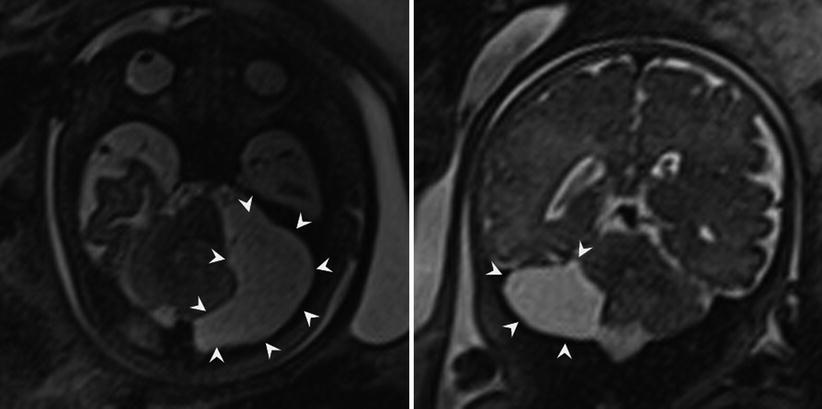
A Blake pouch cyst is an ependymal-lined outpouching of the dorsal and inferior membrane of the fourth ventricle (e.g., inferior medullary velum). This results in upward rotation of a normally developed vermis in the setting of a normal appearance of the tentorium and torcula. Blake pouch cysts are included in the Dandy-Walker continuum by some authors, and when very large, it can be difficult to evaluate the vermian integrity. Mass effect from large Blake pouch cysts is possible but extremely rare, and in isolation, these lesions neither have neurodevelopmental consequences nor require treatment [51].
In addition to Blake pouch cyst, mega cisterna magna and arachnoid cysts of the posterior fossa also have a good prognosis, allowing for appropriate patient counseling following their diagnosis. Mega cisterna magna is abundant retrocerebellar fluid within focally enlarged subarachnoid space, defined as a distance between the inner table of the occiput and the cerebellum as >10 mm after 18 weeks gestational age (Fig. 6.17). Mega cisterna magna is in communication with the ventricular system and can be considered a normal variant [20, 120]. Congenital arachnoid cysts are collections of CSF that arise within duplications of the arachnoid membrane (Fig. 6.18). Arachnoid cysts can produce mass effect on the adjacent cerebellum and vermis and can obstruct CSF flow leading to hydrocephalus. Retrocerebellar arachnoid cysts are considered by some to be part of the Dandy-Walker continuum, making a thorough evaluation for additional cerebral anomaly important when an arachnoid cyst is suspected. When very large, arachnoid cysts are managed with shunt placement in infancy but generally have a good prognosis [3]. Approximately 1/3 of both Blake pouch cysts and mega cisterna magna will resolve in utero [47], and the majority will not require further treatment [42].
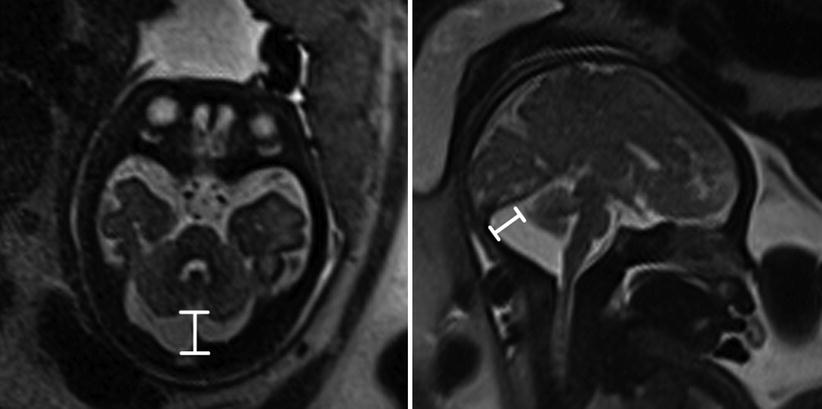
Fig. 6.17
Axial and coronal T2-weighted half Fourier RARE images of a 34-week gestational age fetus with mega cisterna magna (double headed line) measuring 19 mm