Generic name
Trade name
Agent/Target
Cancer indication
Approval
Unconjugated
Rituximab
Rituxan
Chimeric anti-CD20 IgG1
B cell lymphoma
1997
Trastuzumab
Herceptin
Humanized anti-HER2 IgG1
Breast
1998
Alemtuzumab
Campath-1H
Humanized anti-CD52
CLL
2001
Cetuximab
Erbitux
Chimeric anti-EGFR
Colorectal
Head/neck
2004
2006
Bevacizumab
Avastin
Chimeric anti-VEGF
Colorectal
NSCLC
2004
2006
Panitumumab
Vectibix
Human anti-EGFR
Colorectal
2006
Radioconjugates
Satumomab pendetidea, b
OncoScint
111In-murine anti-TAG-72 IgG
Colorectal, ovarian
1992
Nofetumomab merpentana, b
Verluma
99mTc-murine anti-EGP-1 Fab’
SCLC
1996
Arcitumomaba,b
CEA-Scan
99mTc-murine anti-CEA Fab’
Colorectal
1996
Capromab pendetidea
ProstaScint
111In-murine anti-PSMA
Prostate
1996
Ibritumomab tiuxetan
Zevalin
90Y-murine anti-CD20 IgG + rituximab
Follicular B-cell lymphoma
2002
Tositumomab
Bexxar
131I-murine anti-CD20 IgG + unlabeled tositumomab
Follicular B-cell lymphoma
2003
Drug conjugates
Gemtuzumab ozogamicin
Mylotarg
Humanized anti-CD33 IgG4 calicheamicin conjugate
AML
2000
IgG is a large protein of about 150,000 daltons, and composed of pairs of heavy and light polypeptide chains that are assembled to form in a ‘Y’-shaped structure (Fig. 1). Each heavy chain pairs with a light chain to form one “arm” of IgG, and each heavy and light polypeptide chain is divided into heavy and light chains, the former composed of 1 variable and 3 constant domains (VH and CH1, CH2, CH3), and the latter containing 1 variable and 1 constant domain (VL and CL). The composition of the constant regions is highly conserved, while the variable region of the heavy and light chains is highly adaptable, changing in accordance with the antigen that evokes an antibody response. Each of the heavy and light chains responsible for binding to the antigen has three complementarity-determining regions (CDRs) that provide the antibody with its unique structure for binding to the antigen in three dimensions, while the other portion of the variable domain forms the framework that support the CDRs and optimize their binding to antigen. The CDRs are structured to bind with just a complementary site on the antigen known as the epitope. The epitope’s 3-D structure defines its specificity, and depending on the size and complexity of the antigen, multiple unique epitope structures are presented on the antigen molecule to which a unique antibody can be made for binding to the cognate antigen. Immunoglobulins can bind more than one epitope via their multiple “arms,” thereby cross-linking two antigen molecules. In an immune response, antibodies can implement extensive cross-linking, resulting in large lattices of the antigen for improved identification by immune cells (Table 2).
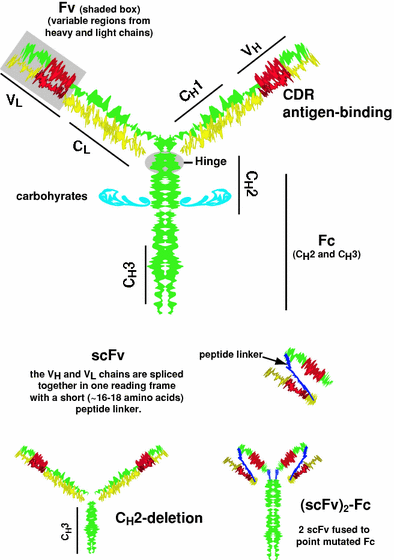
Fig. 1
Schematic model of an IgG and some molecularly engineered antibodies. IgG has 2 heavy (H) and 2 light (L) chains. Disulfide bridges within a hinge region help hold the 2 heavy chains together. Each heavy chain is associated with a light chain. The heavy and light chains are divided into domains, the CH3, CH2, CH1 and VH for the heavy chain and CL and VL for the light chains. Each chain has domains that are highly conserved (constant regions, C), and regions that are more variable (V). Within the variable region of each chain are several highly variable regions known as the complementarity-determining region (CDRs), which form a 3-dimensional structure that binds to the epitope of an antigen. The CH2 and CH3 portion of the IgG are referred to as the Fc-portion, with the CH2 region being glycosylated. The VH and VL regions of the molecule are referred to as the Fv-region. In molecular engineering, the VL and VH regions of the two separate chains are fused in single reading frame, with the two chains tethered together with a 16–18 length polypeptide. This is known as a single chain molecule or scFv (Colcher et al. 1998; Weisser and Hall 2009). One of the earliest of the engineered antibodies modified the heavy chain by deleting the CH2 region. This CH2-deletion form has more rapid clearance from the blood (Mueller et al. 1990; Slavin-Chiorini et al. 1995). Another recently proposed structure fuses two scFvs on an intact Fc, but the Fc contains point mutations to remove its ability to bind to the neonatal receptors that also affect antibody clearance in the blood (Olafsen and Wu 2010)
Table 2
Partial listing of anti-cancer antibodies in advanced clinical testing
Generic name | Agent/target | Cancera |
|---|---|---|
Apolizumab | Human anti-HLA-DR | CLL, NHL |
Chimeric 14.18 | Chimeric anti-ganglioside (GD2) IgG | Neuroblastoma, melanoma |
Cotara | Radiolabeled chimeric anti-TNT | Lung, glioma |
Epratuzumab | Humanized anti-CD22 | NHL |
Galiximab | Humanized anti-CD80 | NHL |
HuMax-CD4 | Fully human anti-CD4 | CTCL |
Lumiliximab | Humanized anti-CD23 (Fc epsilon RII) | CLL |
MDX-010 | Anti-CTLA-4 | Melanoma |
Matuzumab | Humanized anti-EGFR | CRC, NSCLC, pancreatic |
Oregovomab | Murine anti-CA-125 | Ovarian |
Pertuzumab | Humanized anti-HER-2 | Breast, prostate, ovarian |
Rencarex | Chimeric anti-G250 IgG | Kidney |
Vitaxin | Humanized anti-αvβ3 integrin | Melanoma, prostate |
Antibodies are made by plasma cells, derived from B lymphocytes. Normally, an antibody response is polyclonal, meaning the formation of many antibodies each from unique B cells and each reacting with a unique epitope. Experimentally, the clonal population of B cells reactive to an antigen determinant enables the isolation of specific antibody-forming cells (or clones) from the spleen of an immunized animal, such as a mouse. Using the method of ‘hybridization,’ where the immunized B cells are fused with an immortalized myeloma cell line, a hybridoma cell line capable of unlimited production of a monoclonal antibody (MAb) is accomplished. The MAb hybridoma technique, for which Köhler and Milstein received the Nobel prize (Köhler and Milstein 1975), has had a profound impact on the clinical and commercial advance of MAb-based immunotherapy products in several medical indications, as well as on molecular immunology, immunodiagnosis, and clinical laboratory medicine (Sharkey and Goldenberg 2006). For clinical use, murine MAbs have been engineered to substitute human IgG components for murine sequences (Morrison et al. 1984; Jones et al. 1986; Kim et al. 2005; Weiner 2006; Qu et al. 2005). The first of these “de-moused” constructs were chimeric antibodies, where the VL and VH regions of the murine antibody are spliced onto the human IgG, resulting in an IgG that is ~67 % human and 33 % murine. Thereafter, antibodies were further humanized by grafting only the CDRs from the murine MAb, along with some of the surrounding framework regions, onto a human IgG, thus having a molecule with ~5 % of its sequences from the parental murine MAb. More recent advances have resulted in fully human MAbs that are now in clinical practice or trials (Weiner 2006; Moroney 2005). Such engineered MAbs greatly reduce the immunogenicity of antibodies, while permitting the human Fc portion to recruit cellular immunity via complement and effector cells.
While the specificity of antibody binding is determined by its variable regions and CDRs, the heavy chain constant regions also possess important properties. The CH2 and CH3 domains comprise the Fc-portion of the IgG molecule. This portion can bind to Fc-receptor-bearing cells (most notably other immune cells), and is responsible for binding components of the complement cascade. Thus, while one end of an antibody binds to an antigen, for example, on the surface of a foreign cell, the other end, the Fc-portion, can interact with other components of the immune system to attack and destroy this cell. These two attack mechanisms are referred to as antibody–dependent cellular cytotoxicity (ADCC) and antibody-dependent complement–mediated cytotoxicity (CDC) (Fig. 2). ADCC involves the recognition of the antibody by immune cells that engage the antibody-marked cells and either through their direct action, or through the recruitment of other cell types, leads to the tagged-cell’s death. CDC is a process where a cascade of different complement proteins become activated, usually when several IgGs are in close proximity to each other, with one direct outcome being cell lysis, or indirectly is responsible for attracting other immune cells to this location for effector cell function. Today’s molecular engineering can create a myriad of antibody sculptures, tailor-designed with multiple and even varied antigen-binding domains, and with Fc-portions that also have been modified to enhance interactions with other immune cells or alter their pharmacokinetic behavior (Presta 2008; Strohl 2009; Yamane-Ohnuki and Satoh 2009; Olafsen and Wu 2010).
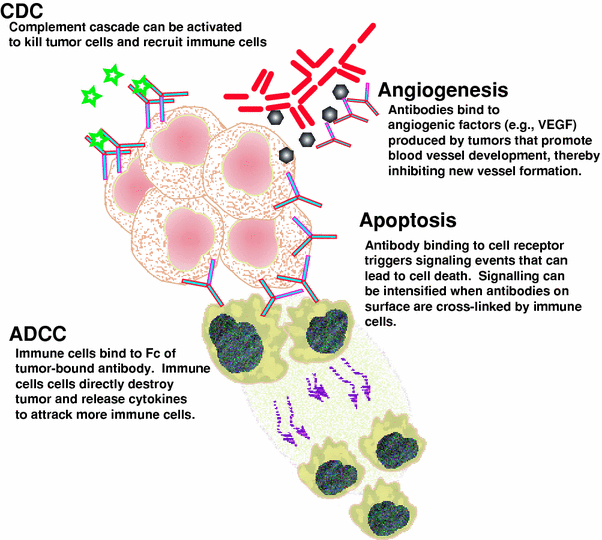
Fig. 2
Schematic representation of the various mechanisms of cell killing by unconjugated antibodies. ADCC and CDC are the more traditional cellular or non-cellular mechanisms that the immune system uses. More recently, we have come to understand that antibodies can disrupt cellular processes, such as sending signals inside the cell that activate apoptosis or they can bind to factors that can impact the growth of tumors. In this example, vascular endothelial growth factor (VEGF) is produced by tumors to stimulate the growth of vascular endothelial cells to provide a blood supply to the tumor
ADCC and CDC are among the most important cellular mechanisms by which antibodies can orchestrate killing of cells or invading organisms. However, antibodies can also interfere with a cell’s metabolic and replicative machinery directly, via attachment to critical receptors involved in these signaling pathways. Some of these signals induce programmed cell death, or apoptosis. One such example involves rituximab, an anti-CD20 MAb that induces apoptosis on target lymphoma cells that express CD20, and is therefore effective in the treatment of B cell lymphomas and leukemias, as well as normal B cells engaged in autoimmune diseases, such as rheumatoid arthritis. When rituximab binds to two CD20 molecules, it triggers CD20 molecules to move within the membrane through lipid rafts, transmitting a signal that initiates a cascade of events that lead to apoptosis (Jazirehi and Bonavida 2005). Studies in vitro have indicated that only a small proportion of the cells actually are killed because of this signaling event, but this level can be increased by crosslinking rituximab with an anti-antibody (Shan et al. 1998; Zhang et al. 2005). Crosslinking can occur in vivo when the Fc-portion of cell-bound antibody is bound by immune cells via their Fc-gamma receptors (FcγR). Since cancer cells usually have alternative, redundant pathways for critical functions, interrupting one signaling pathway might not suffice, so it is not surprising that antibodies affecting different pathways can be combined (Marvin and Zhu 2006; Huang et al. 2009; Quintas-Cardama et al. 2010), and also given to augment the anti-tumor effects of other standard forms of cytotoxic therapy (Jazirehi and Bonavida 2005; Czuczman 2002; Marty et al. 2005; Raben et al. 2005; Chan et al. 2006; Robert et al. 2006), possibly because of sensitizing the target cells to cytotoxic therapies. But, as in the case of rituximab, many of these antibodies of clinical use also have effector functions, and it is still debated whether direct signaling-induced killing or immune mechanisms play the most critical role in immunotherapy with unconjugated MAbs.
3 Immunoconjugates
Antibodies also function as carriers of cytotoxic agents, such as radioisotopes, drugs, and toxins (Fig. 3). In most settings, antibody conjugates are found to be more potent to cancer cells, yet also more toxic to collateral normal tissues, than unconjugated antibodies. In lymphomas, anti-CD20 radioimmunoconjugates have superior anti-tumor activity compared to their unconjugated antibody counterparts, but there is increased concomitant, but manageable, hematological toxicity (Witzig et al. 2002; Davis et al. 2004). The development and prospects of cancer therapy with radiolabeled antibodies, termed radioimmunotherapy, or RAIT (Goldenberg 1978a, b, 1980), will be reviewed.
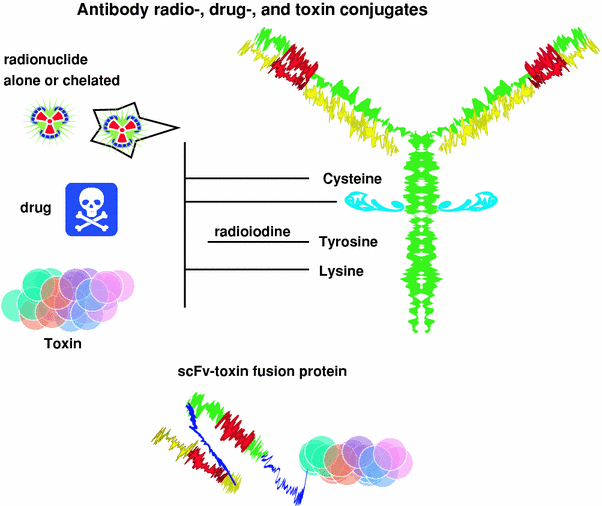
Fig. 3
Antibody conjugates. Radioconjugates and drug conjugates are formed chemically by linking the drug or radionuclide to lysine or by sulfhydryl-linkage chemistry. The radionuclide or drug can be attached directly or to an intermediate, such as a chelate that is used to capture radiometals, or dextran that can be preloaded with multiple drug molecules before being coupled to an antibody. Coupling a drug or radionuclide to the carbohydrate of the antibody ensures the coupling does not interfere with antigen binding, because the carbohydrate is typically far enough away from the antigen-binding portion of the molecule. Toxins had previously been chemically coupled to antibodies, but today, immunotoxins are prepared by recombinant engineering that fuses the toxin molecule to an antibody or one of its engineered fragments, such as a scFv
4 Radioimmunoconjugates
Radiolabeled antibodies were first investigated in the 1950s (Pressman and Keighley 1948; Pressman and Korngold 1953; Bale and Spar 1957; Bale et al. 1955), and laid the foundation for subsequent studies undertaken with human tumor xenografts and human tumor antigens in the 1970s (Bale and Spar 1957; Silverstein 2004). The first human cancer target for radiolabeled antibodies was carcinoembryonic antigen (CEA), which was first disclosed by Gold and Freeman as a specific antigen for gastrointestinal cancers (Gold and Freedman 1965), and resulted also in a serum diagnostic assay (Gold et al. 1978; Thomson et al. 1969). 131I-labeled antibody to CEA provided the first clinical evidence that radioactivity could be selectively delivered to human tumors for cancer detection by external imaging (Goldenberg et al. 1978), and later for human colonic cancer therapy in an experimental model (Goldenberg et al. 1981). Early therapy trials by Order et al. (Order et al. 1981; Ettinger et al. 1982) integrated a relatively low dose of a radiolabeled antibody into a standard chemotherapy and radiation regimen, but this did not disclose the activity of the radiolabeled antibody alone. While the emphasis in clinical studies in the early 1980s was for cancer detection by nuclear imaging, therapy studies with radiolabeled antibodies alone were initiated (Carrasquillo et al. 1984; Montz et al. 1986; Goldenberg 1988). The first clinical milestone in RAIT was the report by DeNardo et al. (1987), showing that radiolabeled antibody therapy alone could elicit responses in patients with B-cell lymphomas. This stimulated the development of other antibodies for the therapy of this radiosensitive neoplasm, ultimately leading to the approval of two radiolabeled anti-CD20 antibodies for the treatment of non-Hodgkin lymphoma (NHL).
External beam radiation delivers high-dose-rate radiation for short bursts divided over several weeks, whereas RAIT is typically given as an intravenous injection, attempting to deliver low-dose radiation to tumor systemically. It can take 1–2 days before a radiolabeled IgG achieves its maximum tumor accretion, with peak uptake usually <0.01 % of the injected dose per gram tumor; however, the radioactivity deposited in the tumor can be detected for several weeks (Sharkey and Goldenberg 2005). The low uptake and slow kinetics of radiolabeled IgG targeting not only means that RAIT delivers a lower dose rate than external beam irradiation, but that it is continually present for a period determined by the physical half-life of the radionuclide and the biological half-life of the antibody in the tumor. This continuous, low-dose-rate radiation exposure can be at least as effective as intermittent, high-dose rate, external irradiation (Buchsbaum and Roberson 1996; Hernandez and Knox 2004).
Since the radiolabeled IgG remains in the blood for an extended time, the highly radiosensitive red bone marrow is continuously exposed to the radiation, causing the dose-limiting myelosuppression typical for RAIT. In the more radiosensitive hematological malignancies, this can elicit significant anti-tumor responses. However, solid tumors, which are more radioresistant, are less affected by such doses. In an attempt to reduce the duration in the blood, the use of smaller forms of the antibodies, such as a F(ab’)2 or Fab’, and more recently, molecularly engineered antibody sub-fragments, have been explored (Fig. 1). These fragments have more favorable pharmacokinetic properties that improve the tumor/blood ratios because of having a more rapid blood clearance (Kenanova and Wu 2006). The fraction of the injected activity delivered to the tumor is lower with an antibody fragment than an IgG, but because hematological toxicity is reduced, higher doses of radioactivity can be administered. While there have been some reports of improved therapeutic responses using smaller-sized antibodies, the smaller forms, <60,000 daltons, clear from the blood by the kidneys, which could lead to renal toxicity, especially with radiometals (Behr et al. 1999, 2000; Buchegger et al. 1996). Thus, while radiolabeled fragments reduce the risk of hematological toxicity, because the radiation dose required to kill tumors is higher than the limit tolerated to the kidney, radiometal-labeled antibody fragments that are unable to sustain a higher level in the tumor than the kidneys may never prove to be effective therapeutics. In contrast, radioiodinated fragments are not retained in the kidneys, and therefore a favorable therapeutic window potentially can be achieved (Sharkey et al. 1990; Kenanova et al. 2007).
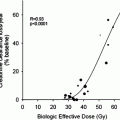
Multi-step pretargeting methods, such as those using bispecific antibodies (discussed in “Radioimmunotherapy of Tumors: Pretargeting with Bispecific Antibodies”), represent a promising new method for imaging and therapy (Sharkey et al. 2005). This involves separating the targeting of the tumor with an antibody from the delivery of radiation either for imaging or for RAIT. The bispecific antibody, which is given in the first step, has at least one arm that binds to the tumor antigen, while a second binds to a hapten. The hapten, which is then given later, is typically incorporated in a small peptide that can be radiolabeled. The unlabeled bispecific antibody given first circulates and binds to the tumor cells, and once it has cleared from the blood and non-targeted tissues, the radiolabeled peptide is given. The small-sized radiolabeled peptide enters the extravascular space very quickly, where it binds to the other arm of the bispecific antibody found on the tumor cells. Within minutes, the non-targeted peptide clears from the blood, typically by urinary excretion within a few hours, while the peptide that localized to the bispecific antibody remains bound to the tumor. This method has been shown in xenograft models to enhance tumor/blood ratios by as much 40-fold, while increasing tumor accretion by as much as 10-fold compared to a directly radiolabeled antibody fragment (Sharkey et al. 2005; McBride et al. 2006). We have found that this pretargeting can increase the total radiation delivered to tumors 1.5-fold, and because uptake occurs so quickly, the dose rate is also increased 3-fold, resulting in improved therapeutic responses (Karacay et al. 2005). Also important is that the small radiolabeled peptide does not have as high accretion in the kidney as found with directly radiolabeled antibody fragments; therefore, tumor/kidney ratios are favorable.
Table 3 lists some of the more commonly used radionuclides bound to antibodies for cancer RAIT. Medium-energy beta-emitters, such as 131I (~0.5 MeV) and 177Lu (~0.8 MeV), traverse ~0.5 to 1.0 mm; high-energy beta-emitters, such as 90Y or 188Re (~2.1 MeV), penetrate up to 11 mm, thus enabling beta-emitters to kill several hundred cells in their path, which is referred to as the bystander, or crossfire effect (Kassis and Adelstein 2005). This is an important attribute of RAIT as compared to other immunoconjugates, since the radioimmunoconjugates can be therapeutic even with heterogeneity of antigen expression. Thus, radionuclide selection is most related to tumor size and extent. Higher energy beta-emitters can kill across a longer path-length, but the absorbed fraction is higher for the lower energy beta-emitters (i.e., probability of hitting the nuclear DNA), making them more efficient for cell destruction. Alpha-emitters, such as 213Bi and 211At, have path lengths of only a few cell diameters, but are also more potent than a low-energy beta particle, requiring fewer “hits” to effect cell killing (Kassis and Adelstein 2005). Low-energy electrons, such as Auger emitters (e.g., 125I, 67Ga, and 111In), have to be in close contact, preferably in the cell or near the nucleus, to be cytotoxic (Mattes 2002). Thus, beta emitters are best used where tumors are ≥0.5 cm in diameter; otherwise, a substantial portion of the energy from the radioactive decay will affect surrounding normal tissues. Alpha and low-energy electron emitters are preferred when there is a small disease burden or with single-cell populations (e.g., leukemia, malignant ascites) (Michel et al. 2003; Kotzerke et al. 2005).
Table 3
Physical properties of several radionuclides used for RAIT
Radionuclide | Emission | Half-life* | Rangea | Approximate # cell diametersb |
|---|---|---|---|---|
90Yttrium | β | 2.7 days | 4.0–11.3 mm | 400–1100 |
188Rhenium | β | 0.7 days | 1.9–10.4 mm | 200–1000 |
131Iodine | β | 8.0 days | 0.08–2.3 mm | 10–230 |
67Copper | β | 2.6 days | 0.05–2.1 mm | 5–210 |
177Lutetium | β | 6.7 days | 0.04–1.8 mm | 4–180 |
211Astatine | α | 7.2 h | 60 μm | 6 |
213Bismuth | α | 0.8 h | 84 μm | 8 |
125Iodine | Auger | 60.5 days | <100 nm | <1 |
111Indium | Auger | 3.0 days | <100 nm | <1 |
5 Radioimmunoconjugate Products
Two radioimmunoconjugates are currently approved in the USA for the treatment of indolent and transformed forms of NHL, 90Y-ibritumomab tiuxetan (Zevalin, Spectrum) and 131I-tositumomab (GlaxoSmithKline) (Sharkey et al. 2005). Both have shown higher objective response rates compared to the unlabeled anti-CD20 antibody used alone (Witzig et al. 2002; Davis et al. 2004). Nevertheless, the pivotal trial comparing 90Y-ibritumomab tiuxetan to rituximab did not show a statistical improvement in the duration of the response, but further follow-up has shown that the complete responses, which were more prevalent with 90Y-ibritumomab tiuxetan, have also been more durable (Gordon et al. 2004a, b). 131I-tositumomab has also shown durable responses, and when used as a front-line therapy, it is better tolerated and may improve responses compared with standard chemotherapy (Fisher et al. 2005; Kaminski et al. 2005; Buchegger et al. 2006). Clinical studies are also evaluating RAIT as a front-line treatment, and that these treatments do not preclude the use of cytotoxic therapies, and in fact may be used in concert as part of a conditioning regimen or consolidation therapy (Bethge et al. 2010; Chatal et al. 2008; Devizzi et al. 2008; Jacobs et al. 2008; Krishnan et al. 2008; Morschhauser et al. 2008; Shimoni et al. 2008; Zhang and Gopal 2008; Zinzani et al. 2008a, b, c; Esmaeli et al. 2009; Hainsworth et al. 2009; Morschhauser et al. 2009; Perrotti et al. 2009; Wang et al. 2009; Burdick et al. 2010; Cicone et al. 2010; Goldsmith 2010; Hoffmann et al. 2010; Hohloch et al. 2010; Kang et al. 2010; Leahy and Turner 2010; Zinzani et al. 2010a, b). However, more randomized clinical trials are needed to assess long-term follow-up and also the risk of late toxicities (e.g., myelodysplasia or leukemia) (Connors 2005). Although there has been some concern about the incidence of late toxicities, there is increasing evidence that RAIT alone might not have been the underlying cause, particularly because many of the patients enrolled on these studies have had extensive prior therapies (Magni et al. 2010; Bennett et al. 2005; Czuczman et al. 2007; Focosi et al. 2008; Friedberg 2008; Link et al. 2010; Roboz et al. 2007; Sacchi et al. 2008). It should be emphasized that, because the red marrow is so sensitive to radiation, these agents are restricted to use in patients with minimal bone marrow involvement and good hematologic status. Indeed, a Phase II trial using 90Y-ibritumomab tiuxetan in chronic lymphocytic leukemia (CLL) concluded RAIT had significant toxicities, preventing it from being able to provide sufficient benefit in this setting (Jain et al. 2009).
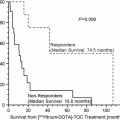
Other radioimmunoconjugates also are being studied in lymphoma and leukemia (DeNardo et al. 1998; Foss et al. 1998; McDevitt et al. 2001; Buchmann et al. 2002; Ma et al. 2002; Zhang et al. 2002, 2006, 2007; Vallera et al. 2003; Glatting et al. 2006) (Table 4). Promising anti-tumor responses have been reported from a Phase I/II trial with a fractionated, weekly dosing of 90Y-epratuzumab tetraxetan (anti-CD22 IgG) in both follicular and other NHL (mantle cell and diffuse large B cell) (Morschhauser et al. 2010). Twelve out of 13 patients (92 %) with follicular NHL who received >30 mCi of the 90Y-antibody in their treatment had a complete response; one had a partial response. The median progression-free survival of this group was 24.6 months, with half still in remission 2–3 years of follow-up. Equally impressive was the fact that 10 of these follicular NHL patients were refractory to a prior anti-CD20 therapy, and 9 of these had complete responses lasting 21.5 months, which is longer than the 6.8-month progression-free survival in rituximab-resistant follicular lymphoma patients given 90Y-ibritumomab tiuxetan (Witzig et al. 2002). The non-follicular group had an overall response of 58 % (11/19), with six being complete or complete/unconfirmed responses, which is also encouraging. Preclinical studies have also suggested a combination of 90Y-anti-CD22 with anti-CD20 antibody therapy may further enhance the clinical response (Mattes et al. 2008). For example, the standard radiolabeled anti-CD20 IgG therapy includes substantial amounts of unlabeled anti-CD20 therapy that is pre-infused before the therapeutic dose of the radioimmunoconjugate, which is given at a relatively low protein dose. While clinical studies concluded that unlabeled anti-CD20 antibody aided tumor localization of the radiolabeled anti-CD20 antibody by blocking the large antigen sink in the spleen or in patients with bulky disease, the rationale for using the currently prescribed amount may be related more to early clinical findings indicting the higher antibody dose enhanced the response even before the therapeutic radioimmunoconjugate was given (Sharkey et al. 2009). Preclinical studies have shown anti-CD20 antibody-induced signaling can enhance the cell’s susceptibility to radiation (Kapadia et al. 2008; Skvortsova et al. 2005, 2006), but also anti-CD20 therapy is itself effective. Thus, combining a radiolabeled anti-CD22 antibody therapy with an anti-CD20 antibody therapy would allow both agents to bind optimally to the tumor, with the anti-CD20 therapy given in advance having the ability to reduce some of the normal B cells that could affect the radiolabeled anti-CD22 targeting, and also initiate signaling events in the tumor that might enhance response.
Table 4
 (Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
 Thyroid Carcinoma
Thyroid Carcinoma
 of Progressive Dedifferentiated and Medullary Thyroid Cancer with Radiolabeled Somatostatin Analogs
of Progressive Dedifferentiated and Medullary Thyroid Cancer with Radiolabeled Somatostatin Analogs
 Bisphosphonates for Bone Pain Palliation
Bisphosphonates for Bone Pain Palliation
 Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
 Use of Dosimetry in the Planning of Patient Therapy
Use of Dosimetry in the Planning of Patient Therapy




Partial listing of radiolabeled anti-cancer antibodies used in clinical testing as listed in clinicaltrials.gov
Related posts:
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
Thyroid Carcinoma
of Progressive Dedifferentiated and Medullary Thyroid Cancer with Radiolabeled Somatostatin Analogs
Bisphosphonates for Bone Pain Palliation
Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
Use of Dosimetry in the Planning of Patient Therapy
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
