Fig. 1
Progress in 68Ge/68Ga radionuclide generators I: design of cows towards medicine
The concept of the solid-phase-based generator is illustrated in Fig. 2; the original sketch is taken from the original publication by Yano and Anger (1964), cf. Fig. 3.
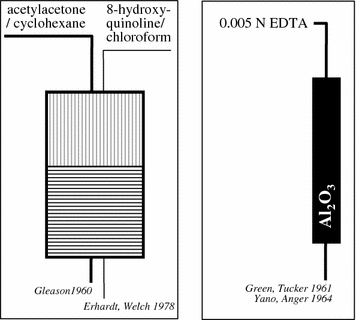
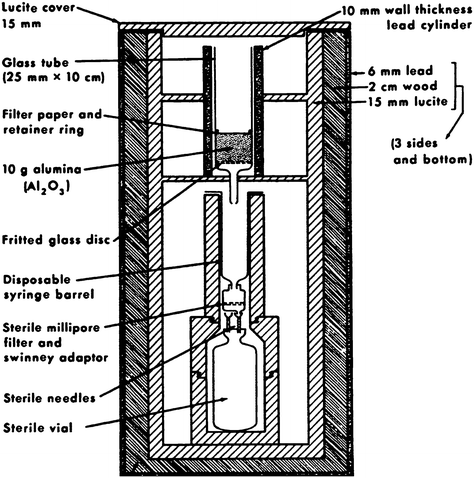
Fig. 2
Progress in 68Ge/68Ga radionuclide generators II: from liquid–liquid extraction to solid-phase-based elution
Fig. 3
Solid-phase-based (alumina) elution (by EDTA solution), taken from the original publication by Yano and Anger (1964)
These solid-phase chromatographic generators offered excellent radiochemical characteristics. Using an alumina column and EDTA as eluent (10 mL 0.005 M EDTA), 68Ga was easily and repeatedly eluted in 95% yield without the need to introduce stable GaIII as carrier.
Gallium 68, the daughter of 270-day germanium 68 …, is obtained by passing edetate solution through an alumina column upon which 68Ge is placed. The edetate becomes labeled with the short half-life 68Ga, while the 68Ge remains on the column to be “milked” again. … Sterility is insured by using a suitable eluting solution and sterilizing the eluent with a Millipore filter (Yano and Anger 1964).
The eluate fraction contained as little as <1.4 × 10−5% of the parent 68Ge. Before injection, 0.5 mL 18% NaCl solution was added to the eluate. This system served as a convenient and economical source of 68Ga-EDTA. Indeed, nuclear medicine physicians such as Gottschalk and others made extensive use of this tracer.
In a way, this radionuclide generator was basically a synthesis unit of a relevant radiopharmaceutical, 68Ga-EDTA (named “veronate” at that time).
2.2 68Ga-EDTA: The PET Pharmaceutical, Development of Positron Scintillation Cameras
Around 1960, radioisotopic brain scanning using a number of agents was being evaluated. Among the most satisfactory were the positron emitters arsenic-74 (as arsenate), copper-64 (as versenate), and mercury-203 (as neohydrin).
In this context, Shealy et al. (1964) concluded the following:
Unfortunately these preparations present some technical difficulties and have not achieved widespread use. In searching for another positron-emitter, it is necessary to find one which for use with present equipment has a half-life of at least one hour, but has a biological half-life of not more than a few days. Further, it should be readily obtainable and incorporable into a chemical form of high specific uptake and of low toxicity. Such an element is Gallium-68 with a half-life of 66 minutes. It is particularly useful because it emits 85 per cent positrons with few non-annihilation gammas. It is a natural decay product of Germanium-68, which has a half-life of 250 days. Such a continuous and relatively inexpensive supply of isotope is desirable if the scanning test is to become widely used. A short-lived isotope such as Ga88 is of particular interest for use with camera-type detection systems. …These show promise of reducing scanning time from the present hours to minutes.
68Ga-EDTA and in a few cases more 68Ga tracers have been adapted for human applications quite quickly by various groups in the USA. (EDTA itself and its relation to CaII were established well known to be nontoxic.) For early applications, see, e.g., Gottschalk and Anger (1964a, b) and Schaer et al. (1965). Systematic application for brain imaging was reported, with medical impact seriously depending on the type of imaging applied. Conventional imaging appeared to by relatively difficult, and relatively high activities of 68Ga-EDTA were needed for valuable medical information. A typical sac of these years is illustrated in Fig. 4.
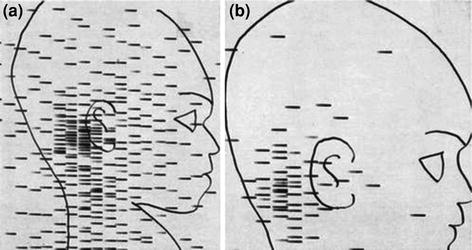
Fig. 4
“Cisternal blockade in an infant with posterior spread of a glioma of the optic chiasm. Five hundred μc of 68Ga were given via the lumbar route. In the first scan (Scan a) an early rise to the cerebellar cisterns is found. Later scans, including Scan b, showed that the isotope remained in the posterior fossa and did not pass upward” (McQueen and Abbassioun 1968)
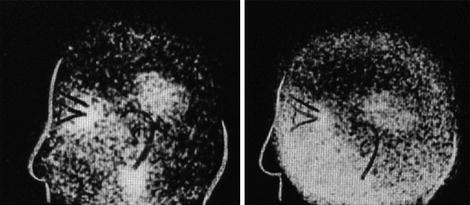
Hal Anger thus started to develop the basics of positron imaging tomography (see, e.g., Anger and Gottschalk 1963; Gottschalk 1996, 2004), arguing as follows (Gottschalk and Anger 1964a, b) (Fig. 5):
… We seriously question whether satisfactory results can be obtained with the conventional positron scanner. Recent phantom studies indicate that the positron scintillation camera using G68 EDTA will detect lesions ½ the volume that can be detected by the conventional positron scanner using As74. The increase in sensitivity is obtained even though the phantom was set up to simulate our clinical condition where brain pictures are obtained in 4–10 minutes with a dose of 350–750 microcuries of Ga68-EDTA. Shealy et al., however, found that 2–3 millicuries of Ga68-EDTA was sometimes an inadequate dose with their positron scanner.
Despite these new features and the great success of 68Ga-EDTA molecular imaging, there was an obvious limitation of the generator application, as it was in practice limited exclusively to synthesis of 68Ga-EDTA. Transfer of 68Ga out of the thermodynamically very stable (log K = 21.7) eluate species 68Ga-EDTA was not straightforward. Yano and Anger (1964) reported, that “attempts are being made… to freed 68Ga… from the EDTA complex.” The procedure, however, was far from user friendly. The time required was given as 30 min, and the transfer yield at 10 mg Ga carrier was 60% (Yano and Anger 1964). The protocol was as follows:
(1) The cow is milked with 10 mL of 0.005 M EDTA solution, and the 68Ga is collected in a 40 mL centrifuge tube. (2) 10–20 mg of carrier GaCl3 in HCl solution is added. (3) The 0.5 mL of saturated ammonium acetate solution is added. (4) Concentrated NH4OH is added drop wise (about 1 mL) to precipitate Ga(OH)3 at pH 6.0. (5) The solution is heated in a boiling water bath for 10 min to coagulate the Ga(OH)3. (6) The solution is centrifuged, and the supernatant solution is discarded. (7) The Ga(OH)3 is dissolved with a minimum volume of hot 20% NaOH. (8) The solution is acidified with about 1 mL of concentrated HCl.
3 Hibernating 68Ga Medical Applications, but New Chemistry Ahead
The impact of 68Ga imaging started to fade away in the late 1970s, mainly because of two reasons: the generator design itself appeared inadequate for the various requirements of 68Ga radiopharmaceutical synthesis, and those available through existing technology had minor clinical relevance, in particular in view of the parallel and rapid developments of the new classes of 99mTc- and 18F-labeled diagnostics.
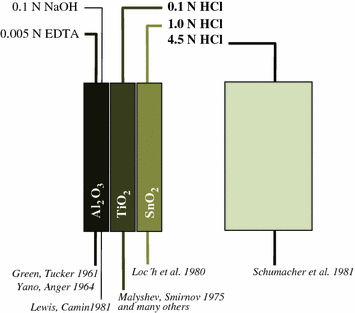
At the same time, however, numerous basic radiochemical papers in the 1970s and 1980s described the use of inorganic matrixes as well as organic resins, selectively adsorbing 68Ge and providing 68Ga desorption within hydrochloric acid solutions of weak (0.1–1.0 N) or strong (>1 N) concentrations, respectively.
In fact, the challenge was to design radiochemical separation systems to provide cationic 68Ga eluate species available for versatile radiolabeling chemistry. Ga(III) exists as cationic species [either pure water-hydrated aquo-complexes such as the hexa-aqua cation Ga(H2O)63+, or similar monochloro or monohydroxo species]. This speciation is easily achieved in solutions of hydrochloric acid with pH ranging between approximately 0 and 2 (i.e., 0.01–1.0 N HCl). For this purpose, MeIVO2-type matrixes (Me = Sn, Ti, Zr, Ce, etc.) appeared to be adequate, effectively adsorbing 68GeIV (e.g., Kopecky et al. 1973, 1974; Malyshev et al. 1975; Loc′h et al. 1980; Ambe 1988). Alternatively, organic resins have been developed, eluting 68Ga in even stronger HCl (e.g., Arino et al. 1978; Neirinckx et al. 1980; Schumacher and Maier-Borst 1981) (Fig. 6).
 via Cation Exchange Cartridges: Versatile Options
via Cation Exchange Cartridges: Versatile Options
 Experience in Peptide Receptor Radionuclide Therapy
Experience in Peptide Receptor Radionuclide Therapy
 Intermediate-Affinity Monoclonal Antibodies Targeting EGFR and HER2/c-neu: Preparation and Preclinical Evaluation
Intermediate-Affinity Monoclonal Antibodies Targeting EGFR and HER2/c-neu: Preparation and Preclinical Evaluation
 Novel 68Ga-Labeled Pteroic Acid-Based PET Tracer for Tumor Imaging via the Folate Receptor
Novel 68Ga-Labeled Pteroic Acid-Based PET Tracer for Tumor Imaging via the Folate Receptor
 Rare Case of a Large Spinal Meningioma with Mediastinal Extension and Malignant Behavior Classified Histologically as Benign
Rare Case of a Large Spinal Meningioma with Mediastinal Extension and Malignant Behavior Classified Histologically as Benign
 Role of 68Ga-Labeled Somatostatin Analogs in the Workup of Patients with NETs: AIIMS Experience
Role of 68Ga-Labeled Somatostatin Analogs in the Workup of Patients with NETs: AIIMS Experience




Related posts:
via Cation Exchange Cartridges: Versatile Options
Experience in Peptide Receptor Radionuclide Therapy
Intermediate-Affinity Monoclonal Antibodies Targeting EGFR and HER2/c-neu: Preparation and Preclinical Evaluation
Novel 68Ga-Labeled Pteroic Acid-Based PET Tracer for Tumor Imaging via the Folate Receptor
Rare Case of a Large Spinal Meningioma with Mediastinal Extension and Malignant Behavior Classified Histologically as Benign
Role of 68Ga-Labeled Somatostatin Analogs in the Workup of Patients with NETs: AIIMS Experience
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
