FIGURE 22.1-1: Schematic diagram of the RH gene locus on chromosome 1 with the four possible genotypes, including the “partial D” genotype phenotypically mimicking RhD-negative. (From Moise KJ Jr. Hemolytic disease of the fetus and newborn. In: Creasy RK, Resnick R, Iams JD, et al, eds. Creasy and Resnik’s Maternal-Fetal Medicine: Principles and Practice. 6th ed. Philadelphia, PA: Saunders; 2009:479.)
PATHOGENESIS
As Levine and Stetson7 first reported in 1939, a breech in the fetal-maternal interface may expose the mother to various fetal red cell antigens, inciting an immunologic response. The antigen is presented to B lymphocytes of the maternal reticuloendothelial system (RES), activating a primary immune response characterized by large-scale immunoglobulin M (IgM) antibody production over the first several weeks. These antibodies are large and do not cross the placenta, posing no risk to the fetus. Following the initial phase, the maternal response shifts to clonal expansion of immunoglobulin G (IgG)-type antibodies and maturation of small resting lymphocytes into memory B lymphocytes targeted against the specific antigen. Memory B lymphocytes can persist in the mother for decades awaiting reappearance of the relevant antigen to differentiate into plasma cells. If the antigen is presented in a subsequent pregnancy, plasma cells can rapidly proliferate and produce large quantities of IgG antibodies that can cross the placenta and bind to fetal red cells. Factors that increase the likelihood of alloimmunization include the volume of fetal-maternal transplacental hemorrhage (0.2 mL or greater), antigen density, gestational age, and ABO-compatibility. Maternal destruction of ABO-incompatible fetal red cells often occurs prior to presentation of other antigens such that the risk of Rh-alloimmunization is reduced from 16% to 2%.10
Once maternal alloantibodies bind fetal red cells, macrophages within the fetal circulation and spleen adhere and lyse the cells. To counter this destruction, the fetus increases plasma erythropoietin, prompting extramedullary erythropoiesis (primarily in the liver) and release of immature erythroblasts.11 The resultant hepatosplenomegaly may alter the portal and venous architecture leading to portal hypertension and decreased hepatic albumin production. Increased umbilical venous pressure and decreased colloid oncotic pressure trigger extravasation of fluid from the fetal vasculature into the fetal and placental interstitial spaces.12 The fetus attempts to compensate and maintain intravascular volume by increasing plasma aldosterone concentration, but this further decreases the fetal osmolality and hematocrit in a feed-forward cycle, ultimately resulting in the visible fetal and placental edema seen with immune hydrops.13
The mechanism underlying fetal anemia and the development of immune hydrops in fetuses affected by Kell-alloimmunization differs from the processes taking place in the Rh-alloimmunized fetus. In vitro studies of hematopoietic progenitor cells from cord blood reveal suppressed growth of the erythroid progenitor cells in the presence of monoclonal anti-Kell antibodies but not anti-Rh(D) antibodies.14 Anti-Kell antibodies cause fetal anemia by inhibiting erythropoiesis at the progenitor-cell level rather than by RES-mediated hemolysis, as seen in Rh-alloimmunization. This finding becomes important in differentiating the diagnostic workup of Kell-mediated alloimmunization from that caused by most of the other blood-group allo-antigens.
DIAGNOSIS
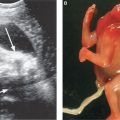
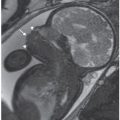
Imaging: Immune hydrops is generally defined by the sonographic detection of excess fluid accumulation within two separate fetal body compartments in conjunction with a positive maternal indirect Coombs test for an antibody known to cause HDFN. Excess fluid accumulation may manifest as ascites, pleural effusion, pericardial effusion, polyhydramnios, skin edema, and placental enlargement. True ascites appears as an echolucent rim of fluid encircling the entire contents of the fetal abdomen such that bowel, liver, and spleen are more easily identified (Fig. 22.1-2). Pleural effusions are often seen bilaterally compressing the lungs and mediastum, potentially leading to pulmonary hypoplasia (Fig. 22.1-3). The diagnosis of pericardial effusion requires visualization of at least 3 mm of fluid surrounding both cardiac ventricles (Fig. 22.1-4). Skin edema must measure 5 mm or greater in thickness (Fig. 22.1-5), and placentomegaly must measure 5 cm or greater in thickness (Fig. 22.1-6). If one such finding is seen in isolation, it should simply be described in terms of its location and not as hydrops fetalis. Although fluid accumulation in two separate fetal compartments denotes hydrops, no sonographic pattern specifies the etiology or severity of anemia.15,16
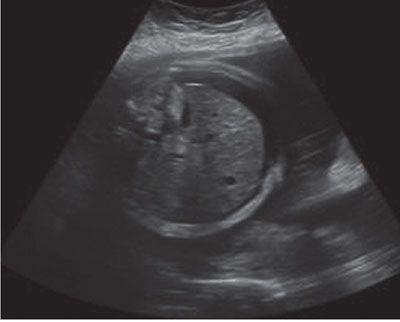
FIGURE 22.1-2: Ascites. Axial image of the fetal abdomen demonstrating ascites.
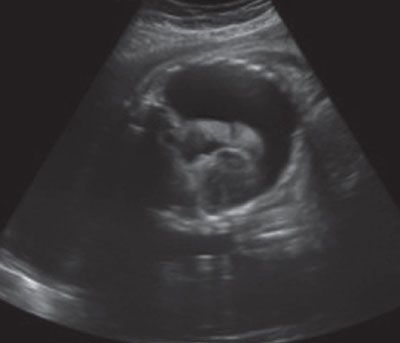
FIGURE 22.1-3: Pleural effusions. Axial image of the fetal thorax demonstrating bilateral pleural effusion with lung compression and mediastinal displacement.
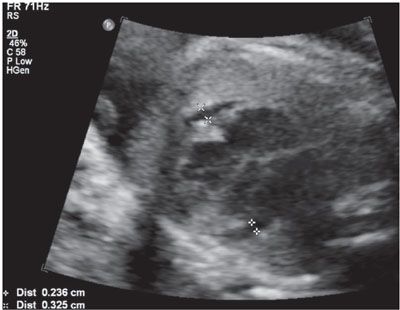
FIGURE 22.1-4: Pericardial effusion. Axial view of the fetal thorax at the level of the four-chamber cardiac view demonstrating more than 3 mm of fluid surrounding both cardiac ventricles.
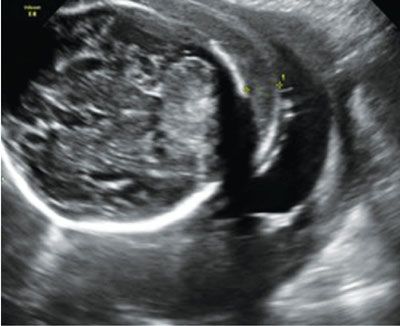
FIGURE 22.1-5: Skin edema of more than 5 mm.
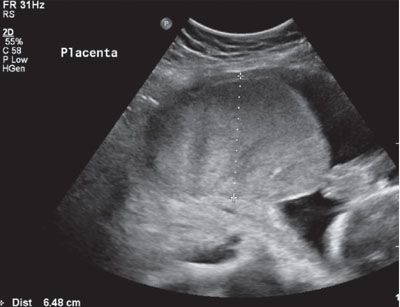
FIGURE 22.1-6: Placental edema greater than 5 cm in thickness.
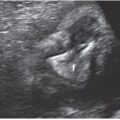
A variety of fetal sonographic measures have been investigated in attempts at identifying methods for noninvasive quantification of fetal anemia. Measurement of the axial splenic circumference (Fig. 22.1-7) demonstrated 100% sensitivity and 94.7% specificity for severe anemia in cases without prior intrauterine transfusion in one prospective study.17 The pathophysiologic association of splenomegaly with hemolytic anemia is easily understood, and this finding was promising. Unfortunately, enlargement of the splenic circumference was not predictive of mild anemia or the decrease in hematocrit following intrauterine transfusion (IUT). To achieve earlier detection and follow-up, investigators have employed Doppler assessment of the fetal blood velocity in the umbilical artery, umbilical vein, descending aorta, ductus venosus, splenic artery, and middle cerebral artery.
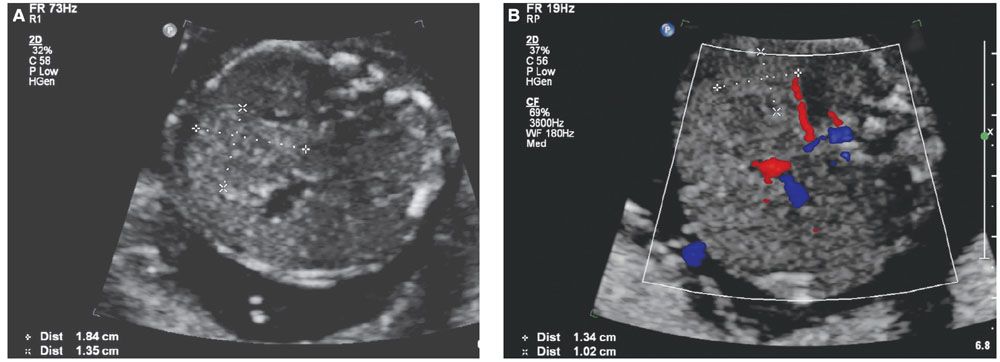
FIGURE 22.1-7: Splenic circumference. A: Axial view of the fetal abdomen at the standard level for abdominal circumference measurement. Splenic circumference (millimeters) = (length + width) × 1.57. B: Color Doppler identifying the splenic artery can aid in location of the spleen.
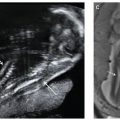
In 2000, Mari and coauthors18 presented convincing evidence supporting the hypothesis that the decreased blood viscosity associated with fetal anemia manifests as an elevation in peak systolic velocity of blood flow within the middle cerebral artery. They prospectively compared cordocentesis-derived hemoglobin concentrations with middle cerebral artery peak systolic velocity measurements in 111 fetuses deemed at-risk for anemia and 265 nonanemic “normal” fetuses. Their findings provided essential normative data and threshold values representing mild, moderate, and severe anemia (see Table 53 in Appendix A1). With appropriate technique (Fig. 22.1-8), this measurement can noninvasively diagnose early anemia prior to the development of hydrops and provide surveillance following IUT to guide timing of repeat percutaneous umbilical cord blood sampling (PUBS) and transfusion. The measurements can be initiated at 18 weeks’ gestation, and the fetal middle cerebral artery closest to the transducer should be evaluated with the Doppler gate placed immediately distal to the bifurcation of the carotid siphon. The angle of insonation should be as close to zero degrees (parallel) to the vessel direction as possible, and the baseline should be adjusted close to zero. Values greater than 1.50 multiples of the median (MoM) for gestational age denote moderate or severe anemia with 100% sensitivity and 88% specificity. The test’s validity is preserved in the cases of Kell-mediated fetal anemia and hydrops.
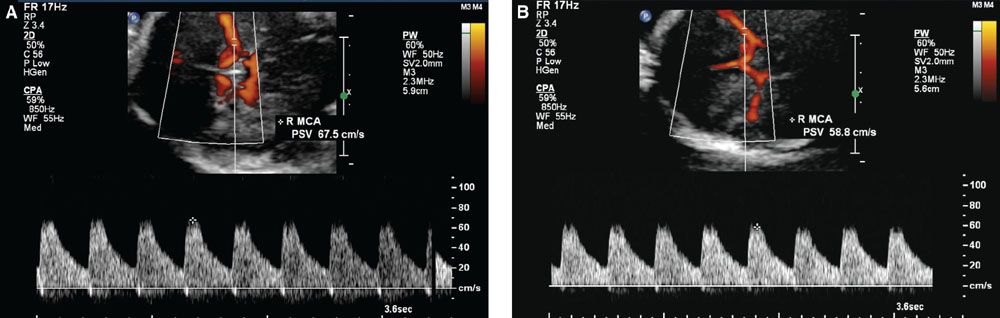
FIGURE 22.1-8: Doppler imaging of the middle cerebral artery. A: Transverse view of the fetal head shows Doppler sampling and peak systolic velocity measurement of the middle cerebral artery with the correct angle of insonation. B: Note the change in the measured value with too large an angle of insonation.
Additional diagnostic tools may be employed for situations where the cause of hydrops fetalis is less clear. Fetal echocardiography may be reserved for nonimmune causes of hydrops. Magnetic resonance imaging may prove useful for investigating brain abnormalities resulting from infectious causes such as congenital parvovirus B19 infection, but its use has been limited to cases of nonimmune hydrops.19
Laboratory Testing: Maternal Rh typing and antibody screening are typically performed in the first trimester, but should be repeated if sonographic evidence of hydrops develops. Although a woman may demonstrate Rh(D) positivity and have a negative first trimester antibody screen, she may acquire atypical blood-group antibodies or become alloimmunized during gestation (following packed red blood cell or platelet transfusion, for example).20 Antibody titers may vary slightly among different laboratories, but most consider titers greater than 1:16 potentially causative of severe fetal anemia. When a maternal antibody screen is positive for non-Kell antibodies and the titer is at or below 1:16, titers are repeated monthly. Maternal Kell antibody titers are not correlative with risk for fetal anemia.14
To determine the likelihood of fetal expression of the suspected antigen, paternal zygosity may be accurately determined by quantitative polymerase chain reaction for the RHD gene or by serology for the other blood-group antigens with a nondominant allele such as the “k” of the (K1) Kell gene.21 Paternal homozygosity essentially confers a 100% likelihood that the fetus is a carrier of the putative blood-group antigen. However, paternal heterozygosity equates to a 50% risk, and determination of fetal antigen status is required. Cell-free fetal DNA testing is now available in the United States such that the exons of the RHD gene are detected with a sensitivity of 97.2% and a specificity of 96.8%.22 This technology has proven reliable for predicting fetal K, C, c, and E phenotypes as well, and these methods are now being used routinely in the United Kingdom but not in the United States.23
Invasive techniques have historically been required to confirm fetal antigen status and quantify anemia as the cause of hydrops fetalis. Amniocentesis accurately determines fetal Rh(D) status with a sensitivity of 98.7% and a specificity of 100%, and it is equally reliable for identifying dominant and nondominant alleles of the other blood-group antigens.24 Amniocentesis for spectrophotometric analysis of amniotic fluid bilirubin levels was first described by Bevis25 and then further characterized by Liley26
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


