Fig. 7.1
Effects of the COMT Val158Met polymorphism on PFC activation. (a) Significant linear effect of the COMT variant on prefrontal cortex activation during a working memory task in 308 healthy controls with increased activation (or decreased efficiency) in Val-allele carriers. (b) At medium working memory load level (2-back task), increasing synaptic dopamine by the administration of amphetamine increases prefrontal efficiency in Val-allele carriers but not Met-allele carriers. Abbreviations: PFC prefrontal cortex (Reprinted with permission from Meyer-Lindenberg and Weinberger (2006) and Tost et al. (2010))
Up to date, an impressive number of single-variant candidate gene approaches have been published in psychiatric imaging genetics, and a full representation of this literature exceeds the scope and possibilities of this chapter. For example, other candidate approaches in the dopaminergic signaling pathway examined risk variants in the dopamine D2 receptor gene (DRD2) and AKT1 (Tan et al. 2008), a gene that plays a crucial role in the D2-dependent noncanonical signaling cascade (AKT1/GSK-3). Risk variants in both genes have been associated with increased risk for schizophrenia (Tan et al. 2008; Cho et al. 2012) and modulation of antipsychotic drug response (Tan et al. 2012; Blasi et al. 2011). Moreover, imaging genetics provided evidence that these variants alter the functional (Tan et al. 2008; Blasi et al. 2011; Bertolino et al. 2010, 2012) and structural (Tan et al. 2008; Markett et al. 2013) properties of frontostriatal circuits in healthy humans. Taken together, these data provide a biological validation of the effects of dopaminergic gene variants in prefronto-neostriatal neural circuits and support the idea for a crucial role of these genetic factors in the pathophysiology and pharmacology of psychosis.
7.3.2 Genome-Wide Supported Risk Variants
Although candidate gene studies offer a principled method to explore promising loci of the genome, these approaches provides only limited support for the pathophysiological relevance of the identified neurogenetic effects if the impact of the genetic variant on the disease phenotype itself is minor (Pezawas and Meyer-Lindenberg 2010). As a consequence, the biological validation of genetic risk variants from genome-wide association studies (GWAS) has gained much popularity because here, the link between genetic variation and categorical diagnosis has been established beforehand at stringent statistical thresholds. On the downside, prior knowledge on the functional role of the identified GWAS variants is typically sparse, an empirical void that can be filled in part by imaging genetics. To date, several studies have identified effects (or the lack thereof (Tost et al. 2010; Wei et al. 2013; Cousijn et al. 2012) of GWAS risk variants for schizophrenia and/or bipolar disorders on structural and functional neuroimaging phenotypes, in particularly for risk variants in the genes ZNF804A (Rasetti et al. 2011; Esslinger et al. 2009, 2011; Thurin et al. 2013; Linden et al. 2013), CACN1AC (Erk et al. 2010; Bigos et al. 2010; Wessa et al. 2010), RELN (Tost et al. 2010), and ANK3 (Linke et al. 2012).
The first such study concerned PFC functional coupling and its modulation by rs1344706A, a genetic variant in ZNF804A conferring risk for schizophrenia and possibly also bipolar disorder (O’Donovan et al. 2008, 2009). The gene maps to chromosome 2q32.1 and encodes for the zinc finger protein 804A, which is abundantly expressed in the developing neocortex and hippocampus (Johnson et al. 2009). Here, connectivity phenotypes derived from functional magnetic resonance imaging (fMRI) are of high interest, as modern neurodevelopmental theories (O’Donovan et al. 2008; Johnson et al. 2009) of schizophrenia conceptualize the disorder as a neural “disconnection syndrome” (Friston and Frith 1995; Stephan et al. 2006; Meyer-Lindenberg 2009), in which genetic and environmental risk factors disturb the formation (and subsequently also the functional interaction and experience-dependent plasticity) of neural regulatory circuits connecting the PFC and hippocampus. Among the available imaging phenotypes, PFC-hippocampus functional coupling during fMRI working memory is the best established intermediate connectivity risk phenotype to date, owing to its established quality criteria (Bilek et al. 2013) and the robust alterations observed in schizophrenia patients (Rasetti et al. 2011; Meyer-Lindenberg et al. 2005), their unaffected first-grade relatives (Rasetti et al. 2011), and genetic animal models of the disorder (Sigurdsson et al. 2010). Using this phenotype, subjects at genetic risk displayed gene dose-dependent alterations in the functional coupling of the PFC and hippocampus (Esslinger et al. 2009) (Fig. 7.2a, b) that mirror the coupling deficits seen in schizophrenia patients (Meyer-Lindenberg et al. 2005). This finding has since been replicated in a large independent sample (Rasetti et al. 2011) and extended (Esslinger et al. 2011) by demonstrating that the ZNF804A coupling effect is cognitive state dependent (i.e., present during working memory function but not during emotion processing or at rest).
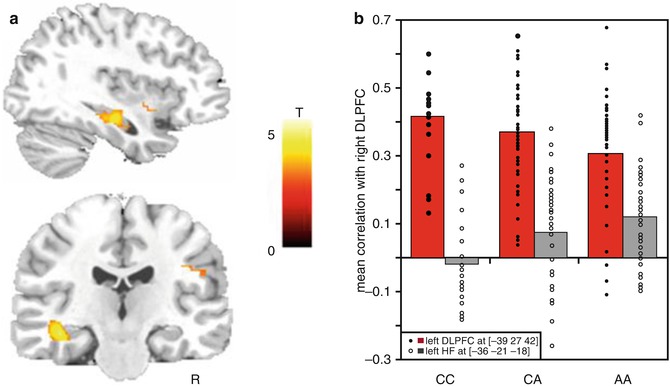
Fig. 7.2
A genome-wide supported variant in ZNF804A modulates PFC functional coupling. (a) Brain regions in temporal lobe where genotype predicts increased correlation with right PFC. (b) Correlation coefficients reflecting connectivity with right DLPFC by genotype: connectivity with left DLPFC shown as red columns and solid diamonds, connectivity with left HF as gray columns and open diamonds. Abbreviations: PFC prefrontal cortex, HF hippocampus formation (Reprinted with permission from Esslinger et al. (2009))
Interestingly, we (Esslinger et al. 2009) and others (Rasetti et al. 2011) have recently shown that healthy carriers of the rs1344706 A-allele do not exhibit differences in PFC regional activation. Moreover, altered PFC-hippocampus connectivity (likely reflecting disturbed plasticity) has been shown to be statistically independent from the well-known fMRI activation or “PFC inefficiency” phenotype (likely reflecting disturbed dopamine release) (Rasetti et al. 2011). Consistent with this, an own recent study (Bilek et al. 2013) has demonstrated that PFC-hippocampus functional connectivity during working memory performance is modifiable by repetitive transcranial magnetic stimulation (rTMS), a plasticity-enhancing intervention that did not impact other neuroimaging phenotypes (i.e., PFC efficiency during working memory, PFC-hippocampus resting-state connectivity). Taken together, these data provide first direct evidence for the “nonredundancy” of different intermediate phenotypes in imaging genetics and point to dissociable biological mechanisms of risk genes at the neural systems level, some of which are promising targets for novel therapies aiming at the modulation of compromised prefrontal network dynamics in schizophrenia (Bilek et al. 2013).
7.3.3 Rare High-Risk Variants
One important lesson from GWAS is the increased occurrence of structural genetic variations (i.e., microdeletions or microduplications) in schizophrenia. Of these, only a copy number variant at 22q11, causing velocardiofacial or 22q11 syndrome, was previously known. These newly discovered microdeletions are associated with a range of brain phenotypes such as schizophrenia but also mental disability, epilepsy, attention-deficit/hyperactivity disorder, and autism (Ben-Shachar et al. 2009). Despite their relative rarity and the complexity of the associated phenotypes, identifying and characterizing structural variations holds considerable potential because they are well-defined genetic alterations with a very high associated risk, which often exceeds that from common genetic variants by more than an order of magnitude (Karayiorgou et al. 2010). While none of the newly identified CNVs have been characterized on the neural systems level, previous work on the 22q11 microdeletion (Karayiorgou et al. 2010) shows that a multimodal imaging approach is feasible and holds the promise to identify neural abnormalities associated with high genetic risk. Further, since it appears likely that copy number variant risk is unlikely to be explained by deletion or duplication of single genes, but rather interactions of genes jointly affected in their expression (Meechan et al. 2007), imaging genetics can ask whether variants in such genes converge on neural systems implicated in schizophrenia. An example of this is the 22q11 microdeletion, which includes, besides COMT and several other candidate genes for schizophrenia, the gene PRODH encoding proline oxidase. A recent study showed that schizophrenia-associated functional polymorphisms in PRODH impact the structure, function, and connectivity of striatum and prefrontal cortex (Kempf et al. 2008). As more and more of these copy number variants are discovered and larger imaging genetics samples permit the study of reasonably sized groups of carriers even for these rare variants, the application of neuroimaging should be very informative in defining neural mechanisms for these high-risk states.
7.4 Accounting for Genetic Diversity
7.4.1 Epistasis
The genetic risk architecture of psychiatric illness is shaped by a large number of risk alleles that are distributed over 3.2 billion base pairs and about 25,000 genes and interact with each other in a largely unknown but surely complex fashion. One aspect of this complexity is captured by the term epistasis, which means that the biological expression of a gene is altered in the presence of one or several other modifying genes. Epistasis is of high relevance to pathophysiology, as seemingly inconsistent association findings for single variants may in fact result from the interaction with other risk factors, including other genes. To date, even larger research consortiums have focused on the examination of pre-hypothesized gene-gene interactions in candidate genes (e.g., “Epistasis Project” on Alzheimer’s disease (Lehmann et al. 2012; Combarros et al. 2009). This relates to the enormous number of potential combinations of genetic risk variants and the resulting high statistical threshold of forward-genetics approaches that exceed the statistical power of even the most comprehensive GWAS to date by far.
Analogous to molecular genetics, research strategies for epistatic interactions of candidate genes have been implemented in imaging genetics. Here, the resulting increase in the number of genotype cells and the multitude of potential confounding factors call for exceptionally careful study designs and the availability of very large neuroimaging databases, which may explain the relative paucity of published imaging epistasis work to date (Tost and Meyer-Lindenberg 2011). A particularly good example for the success of this strategy is a prior study (Nixon et al. 2011) on the effects of the interaction of COMT Val158Met polymorphism and a risk variant (M24) in the d-amino-acid oxidase activator (DAOA) gene. Similar to COMT, DAOA modulates prefrontal functional efficacy during working memory performance (Goldberg et al. 2006) and has been implicated in schizophrenia risk (Chumakov et al. 2002) through epistatic interaction with COMT (Nicodemus et al. 2007), possibly by its detrimental effects on N-methyl-d-aspartate receptor function (Chumakov et al. 2002) and downstream alterations in the glutamate-dopamine balance of frontal-striatal networks (Tost and Meyer-Lindenberg 2011).
The carefully designed study (Nixon et al. 2011) demonstrated significant effects of DAOA by COMT epistasis on prefrontal efficacy during working memory, as indicated by a disproportionately higher PFC activation (or decreased efficiency) in carriers of both deleterious alleles (i.e., DAOA T/T and COMT Val/Val) at a fixed level of performance. In the context of COMT Met, however, efficient PFC signaling was seen in DAOA T/T carriers. The study is compelling, as it provides a functional validation of a previously reported statistical epistasis of DAOA and COMT on a well-established intermediate phenotype linked to the genetic risk for schizophrenia. Other epistasis studies on dopamine- and glutamate-regulating genes in PFC demonstrated complex functional effects COMT Val158Met in interaction with risk variants in AKT1 (Tan et al. 2008), the dopamine transporter gene (DAT, 3′ VNTR) (Bertolino et al. 2008), and the type II metabotropic glutamate receptor 3 gene (GRM3) (Tan et al. 2007). Taken together, these findings highlight the nonadditive nature of genetic architecture of the brain and draw attention to the fact that imaging genetics, meaningful signals may be easily overlooked if the research scope is restricted to the examination of the main effects of variants in single genes or their linear combination.
7.4.2 Haplotypes
Haplotype analyses go one step further in characterizing the genetic complexity that can be addressed with imaging genetics (Consortium 2005). The term refers to a set of statistically associated SNPs at adjacent loci of the chromosome that reflects the linkage disequilibrium structure of genetic variants in a given population (Hinds et al. 2005). Compared to single alleles, haplotypes may show stronger associations with genetically complex disorders as they contain more information about the underlying causal variants. The study of haplotypes is particularly important when several functional variants are implicated within a single gene or haplotype block, as here, the net effect on brain biology will be critically determined by the specific constellation of coexisting alleles.
The COMT gene is again a very good example. Here, prior evidence for a disease association of the Val158Met coding variant (rs4680) alone has been conflicting (Fan et al. 2005). Also, multiple functional variants have been described in COMT that impact prefrontal dopamine signaling and show association with schizophrenia risk (Bray et al. 2003; Shifman et al. 2002) (e.g., rs2097603 in the P2 promoter (Chen et al. 2004), rs165599 in the 3′ region (Bray et al. 2003)). Two prior imaging genetics studies built upon this prior knowledge and tested whether complex genetic variation in COMT modulates brain function (Meyer-Lindenberg et al. 2006) and structure (Honea et al. 2009) in a pattern that is consistent with interacting functional variants and developed a general methodological solution to the problem that haplotypes are usually only known probabilistically. Among others, these studies demonstrated that a 2-SNP haplotype composed of rs4680 (G/A) and rs2097603 (A/G) showed associations with hippocampus volume (Honea et al. 2009) and prefrontal functional efficiency during working memory (Meyer-Lindenberg et al. 2006) in a pattern that is consistent with a nonlinear (inverted U-shaped) effect of extracellular dopamine on these schizophrenia-related phenotypes.
Another multimodal imaging genetics haplotype study focused on PPP1R1B (Meyer-Lindenberg et al. 2007). The gene maps to chromosome 17q12 and encodes for the dopamine-regulated and cyclic adenosine 3′,5′-monophosphate-regulated phosphoprotein of molecular weight 32,000 (DARPP-32), which is expressed in the striatum and integrates dopamine neurotransmission with the signals from other transmitter pathways (e.g., glutamate, serotonin, neuropeptides) (Svenningsson et al. 2003, 2004). The multimodal neuroimaging study (Meyer-Lindenberg et al. 2007) identified a frequent 7-SNP PPP1R1B haplotype (CGCACTC) that was associated with schizophrenia risk in a family-based association analysis and the expression of PPP1R1B isoforms in postmortem human brain (Meyer-Lindenberg et al. 2007). Moreover, the risk haplotype modulated cognitive performance, striatal gray matter volume, striatal activation during working memory and emotion processing, as well as striatal functional connectivity to the PFC in a large sample of healthy volunteers (Fig. 7.3) (Meyer-Lindenberg et al. 2007). These data demonstrate the value of haplotype analysis in imaging genetics to identify complex and nonadditive genetic interactions in candidate risk genes in frontostriatal regulatory systems and highlight their potential role in the pathogenesis of schizophrenia.
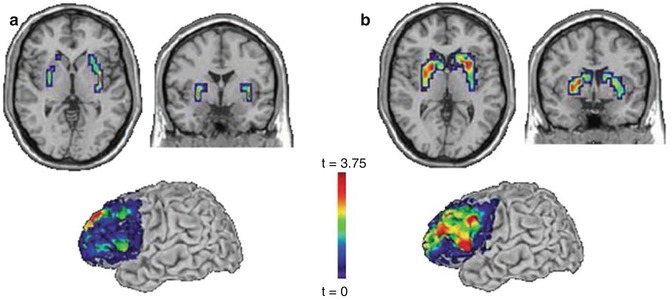
Fig. 7.3
Effects of a PPP1R1B haplotype on frontostriatal activation and connectivity. (a) Working memory: significantly reduced activity in putamen (top) and greater functional connectivity between prefrontal cortex and striatum (bottom) for homozygotes for the frequent (CGCACTC) haplotype. (b) Emotional face matching: significantly reduced reactivity in striatum (top) and greater functional connectivity between prefrontal cortex and striatum (bottom) for homozygotes for the frequent (CGCACTC) haplotype (Reprinted with permission from Meyer-Lindenberg et al. (2007))
7.4.3 Genome-Wide Strategies on Imaging Phenotypes
An original hope of the endophenotype concept was to use these quantitative traits to identify new genes that are relevant for mental illness (Gottesman and Gould 2003). Lately, the broad availability of high-throughput genotyping in combination with ever-larger imaging datasets has made this possible in imaging genetics. For example, the imaging GWAS approach has been advanced by large multinational consortia such as the Enhancing Neuro Imaging Genetics Through Meta-Analysis (ENIGMA) (The ENIGMA Network 2010) or Alzheimer’s Disease Neuroimaging Initiative (ADNI) (Mueller et al. 2005) work groups, which seek to share databases and analysis strategies to attain sufficiently powered cohorts and aid the replication of neurogenetic association findings. Common outcome measures are well-established structural intermediate phenotypes (e.g., hippocampus volume (Peper et al. 2007; Kremen et al. 2010)) derived from the spatial reduction of high-resolution 3D MRI datasets, for example, by calculating summary measures over automated anatomical parcellations. A high-profiled imaging GWAS study (Stein et al. 2012) included over 21,000 datasets and identified a very strong (P = 6.7 × 10−16) and genome-wide significant (P < 1.25 × 10−8) association of an intergenic SNP (rs7294919) at 12q24.22 with bilateral hippocampus volume. Using three independent samples of human brain tissue, the study also provided evidence for a functional role of the variant by demonstrating converging effects of rs7294919 (or SNPs in allelic identity) on the expression of TESC (Stein et al. 2012), a positional candidate gene involved in cell proliferation and differentiation during neurodevelopment (Bao et al. 2009).
A conceptual extension of imaging GWAS, voxel-wise GWAS (vGWAS), identifies associations between genetic variants and neuroimaging phenotypes in a whole-brain whole-genome approach, i.e., without a priori limitation of the genetic survey to spatially circumscribed neural regions of interest. An advantage of vGWAS over is that it offers greater anatomical detail of the detected genomic associations, thereby potentially increasing the statistical power of the approach. The first published vGWAS study (Stein et al. 2010) in the field quantified individual differences in brain structure with tensor-based morphometry and explored 448,293 SNPs for associations with 31,622 brain voxels in a sample of 740 elderly subjects with high-resolution structural MRI data from the ADNI consortium. Here, only the P value of the most highly associated (“winning”) variant was retained for each voxel, and multiple comparison correction across SNPs and voxels was achieved by modeling the distribution of minimum P values under the null hypothesis and subsequent false discovery rate (FDR) correction of the corrected P-maps. Although no variant was detected that survived the strict significance criterion, several SNPs were identified that merit further investigation such as a variant in CADPS2, a gene involved in neuronal monoamine uptake that was associated with brain structure in the lateral temporal lobe (rs2429582, 7q31.32, uncorrected P min = 4.23 × 10−10) (Stein et al. 2010).
Several other genome-wide strategies are currently in translation from molecular genetics to imaging genetics. Among others, this includes the association of polygenic risk scores with established neuroimaging intermediate phenotypes, a method initially applied by the International Schizophrenia Consortium (ISC) (Purcell et al. 2009) to test the polygenic theory of psychiatric disorders (Gottesman and Shields 1967), which postulates that numerous common genetic risk variants with small effect sizes have an important biological role in combination (Wray et al. 2007). Here, GWAS “training datasets” are used to identify a very large number of putative risk alleles distributed across the genome that are associated with an established schizophrenia intermediate phenotype at very relaxed thresholds (e.g., P <0.5). In subsequent independent “test datasets,” individual polygenic risk scores are assigned, based upon the cumulative number of putative risk alleles and their weighted effect sizes, and subsequently tested for associations with the imaging phenotype. Another method relevant to imaging genetics is gene-set enrichment analysis (GSEA) (Potkin et al. 2010). The approach takes advantage of the fact that common genetic risk variants for psychiatric disorders lie among sets of genes with overlapping functions. By combining whole-genome genotype information, neuroimaging, and a priori knowledge of empirical databases on the genetic contributions to molecular functions or biological processes, GSEA tests whether variants, which are more strongly associated with a given imaging intermediate phenotype of interest, tend to significantly aggregate in “gene sets” and biological pathways that are of relevance to the illness (Mootha et al. 2003; Subramanian et al. 2005).
7.5 The Role of the Environment
Consistent with earlier (Gottesman and Shields 1972; McEwen and Stellar 1993) and contemporary (McEwen 2012; Booij et al. 2013) accounts on the diathesis-stress model, adverse environmental factors clearly contribute to psychiatric disorders such as schizophrenia, notwithstanding the fact that they are highly heritable (van Os et al. 2010). Many of these factors (e.g., childhood adversity) tend to increase the risk for mental illness and other unfavorable developmental outcomes in a rather unspecific fashion. However, two stressors linked to the social environment have been primarily associated with schizophrenia, urban upbringing and migration. Prior epidemiological research has repeatedly demonstrated that there is at least a twofold increase in schizophrenia risk in individuals that have been raised in urban environments (van Os et al. 2005; Krabbendam and van Os 2005) or belong to ethnic minority populations (van Os et al. 2010; Cantor-Graae and Selten 2005; Bourque et al. 2011). The adverse effects of urbanicity and migration have been established in various countries and ethnicities worldwide, are particularly detrimental when the exposure occurs in childhood and early adolescence (Krabbendam and van Os 2005; Veling et al. 2011), and interact with genetic risk factors for the illness (Krabbendam and van Os 2005; van Os et al. 2008). These observations, especially the likely early impact of these risk factors, are consistent with the proposed neurodevelopmental origin of the disorder (Weinberger 1987) and suggest that the pathway to psychosis is, at least in parts, socioneurodevelopmental in nature (Morgan et al. 2010). A commonly proposed intermediate of the association of social risk factors and psychosis is chronic social stress induced by, for example, repeated exposure to social fragmentation (urbanicity) or racial discrimination (migration) in early life. This may impact the functional organization of neural regulatory circuits during development (Niwa et al. 2013; Burghy et al. 2012; McEwen 2012) and dispose the brain to abnormal stress responses and psychotic experiences in adulthood (see Fig. 7.4) (van Os et al. 2010; Cantor-Graae and Selten 2005; Bourque et al. 2011; Selten and Cantor-Graae 2005).
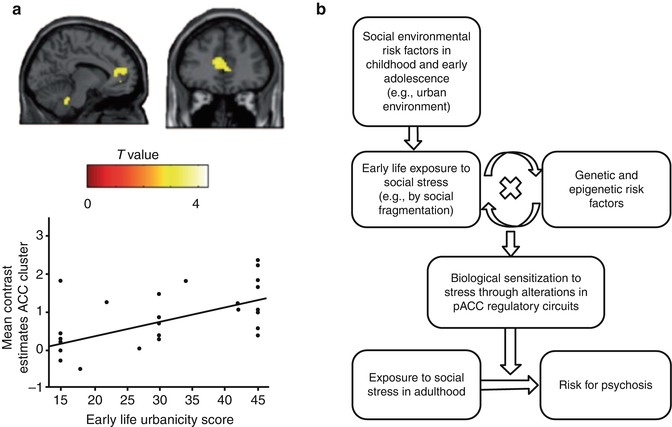
Fig. 7.4
Neural mechanisms of social environmental risk factors. (a) Relationship between urban upbringing and pACC function in humans. Individuals raised in urban environments show increased brain response to social evaluative stress in pACC. (b) Specification of the diathesis-stress model to the risk factor urban upbringing: the model proposes that early exposure to social stress and the adverse interaction with genetic and epigenetic risk factors result in a sensitization of disease-related neural regulatory circuits. This may lead to abnormal neural stress responses in adulthood and promote the manifestation of psychotic phenomena. Abbreviations: pACC perigenual cingulate cortex (Reprinted with permission from Lederbogen et al. (2011) and Haddad and Meyer-Lindenberg (2012))
Only recently, neuroimaging studies have begun to identify neural mechanisms that mediate the effects of social environmental risk factors such as urban upbringing (Lederbogen et al. 2011) or social status (Zink et al. 2008; Gianaros et al. 2007) on the integrity of neural regulatory circuits (Meyer-Lindenberg and Tost 2012; Tost and Meyer-Lindenberg 2012). These initial data point to a vulnerability of the perigenual anterior cingulate cortex (pACC), a key region for regulation of limbic activity and negative emotion, to adverse experiences in the social environment. Interestingly, neural abnormalities in ACC have also been established in schizophrenia patients (Radua et al. 2012; Sambataro et al. 2013), individuals at high genetic risk for the disorder (Sambataro et al. 2013), and healthy carriers of genome-wide supported psychosis risk variants in CACNA1C (Erk et al. 2010) or ZNF804A (Thurin et al. 2013). The same area was also prominently implicated in a recent meta-analysis of functional and structural alterations in early schizophrenia (Radua et al. 2012). These observations support prior epidemiological evidence for interacting genetic and environmental susceptibility, suggest a specific neural system where the effects of genetic and social environmental risk factors may converge, and underscore the potential of neuroimaging to interrogate the neural basis of both genetic and environmental risk constellations. The same set of data also motivates an interest in the adverse interaction of both sources of risk. For example, it is well-known that the individual susceptibility to social adversity is moderated by functional polymorphisms in the promoter regions of the serotonin transporter (SLC6A4) (Caspi et al. 2003) and monoamine oxidase A (MAOA) (Kim-Cohen et al. 2006) genes. Thus, future successful attempts of unraveling the sociogenetic risk architecture of mental illness with neuroimaging will require a substantial extension of established imaging genetics approaches to include nongenetic factors and state-of-the-art methods of epigenetics, environics, epidemiology, and social psychology (Meyer-Lindenberg and Tost 2012; Tost and Meyer-Lindenberg 2012).
7.6 Outlook
Since its first application (Heinz et al. 2000) more than a decade ago, the combination of modern neuroimaging methods and genetic mapping techniques has evolved to a successful neuroscience field that utilizes a variety of newly developed research methods to uncover the biological pathways that lead from risk genes over neural circuit abnormalities to psychopathology (Pezawas and Meyer-Lindenberg 2010; Meyer-Lindenberg 2010). To date, imaging genetics has identified several intermediate phenotypes that are reliably altered in healthy individuals at genetic risk and patient populations, thereby identifying relevant pathophysiological processes and providing a biological confirmation of prior molecular genetic associations. As exemplified by the altered activation and functional connectivity of the DLPFC in the context of schizophrenia, this work has already prompted subsequent successful bench work aiming to validate the effects of established and novel treatment strategies for mental illness (Bilek et al. 2013; Blasi et al. 2011; Rasetti et al. 2010; Apud et al. 2007). While imaging genetics has thus proven to be very fruitful, the true potential of the field has not been fully exploited yet, and several remaining challenges need to addressed in order to prompt true clinical innovations in the years to come (Pezawas and Meyer-Lindenberg 2010; Meyer-Lindenberg 2010a, b).
Current limitations include, among others, the predominance of single-variant approaches that fail to account for the true genetic complexity of psychiatric disorders, the high proportion of single-site studies not powered to detect small effect sizes, the relative lack of coordinated attempts for independent replication of association findings, the small amount of translational approaches designed to exploit the benefits of both human neuroimaging and animal research, and the remaining gaps in knowledge on many of the proposed neuroimaging “intermediate phenotypes” in use (e.g., unspecified task quality criteria, inconsistent directionality of effects, unknown link to the genetic risk for disorders, biological redundancy vs. independence of different phenotypes, etc.) (Rasetti and Weinberger 2011




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
