Fig. 1
List of radioprotection recommendations, we give our patients before treatment. In particular, we invite the patient to avoid close contacts with babies and pregnant women, and to pay attention when using the toilet
After informing the patient both verbally and in writing, the patient must give consent to the treatment.
2 Facility and Personnel
1.
Therapy with Zevalin® should only be performed in Nuclear Medicine Departments that meet the standards for treatment with unsealed radioactive sources and licensed according to the national and local regulations.
2.
The personnel involved in the procedures must have the required qualification and the appropriate government authorisation for the use and manipulation of radionuclides.
3.
A NM Physician is responsible for the organisation and the coordination of all pre-treatment and treatment steps along with the referring haematology-oncology service and the radiolabelling procedures.
4.
Proof of training in radiochemical labelling procedures, including quality control, is required.
5.
Zevalin® may be received, used and administered only by authorised persons in designated settings. Its receipt, storage, use, transfer and disposal are subject to national regulations.
3 Treatment Plan
3.1 Patient Data
Patients enrolled in high activity RIT trials are usually affected by histologically confirmed, resistant or refractory CD20 positive, aggressive B cell NHL, not suitable to receive HDCT, as age over 65 years, or in case of relapse or progression after a previous autologous transplantation. (See Fig. 2).
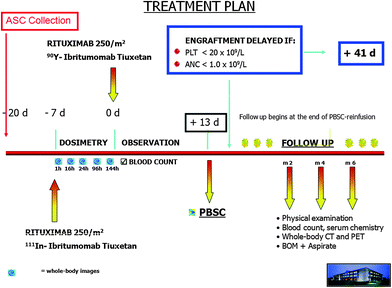
Fig. 2
Treatment plan. Patients receive 111In-Zevalin on day −7 followed by a therapeutic activity of 90Y-Ibritumomab-tiuxetan on day 0. Both administrations are preceded by an infusion of the unlabelled antibody Rituximab (250 mg/m2), to optimise the biodistribution. Following administration of 111In-Zevalin, serial anterior/posterior whole body scans are acquired at 0, 1, 16, 24, 96 and 144 h after infusion to evaluate the distribution of activity in critical organs. Urine and blood samples are also collected at the same timing. Reinfusion is performed on day 13. CT (Computed Tomography) scan, PET (Positron Emission Tomography) scan; BM, bone marrow; SQM, square metre; PLT, platelet count; ANC, absolute neutrophil count
In order to be enrolled in high activity Zevalin® trials, patients must satisfy inclusion criteria, as age >18 years, adequate cardiac (cardiac ejection fraction >50 % by echocardiogram), pulmonary (FEV1 >65 % of predicted or a DLCO ≥50 % of predicted), renal and liver functions (serum creatinine level <2.0 mg/dL, total bilirubin level <2.5 mg/dL, AST/ALT level <4 times upper limit of normal); performance status (PS) ≤2 according to the World Health Organisation scale (Oken et al. 1982) and life expectancy >3 months. Platelets (PLT) must be ≥100 × 109/L; no fixed limit for white blood cells (WBC) and haemoglobin (HB).
Exclusion criteria are a previous administration of RIT or history of human anti-murine antibodies (HAMA).
Moreover, it is mandatory to collect also the following information:
1.
Patient data: age, sex, height, weight, diagnosis.
2.
Indication for the therapy.
3.
Information on the previous therapies (including data on previously completed radiotherapy and/or autologous/allogenic stem cell transplantation). Previous chemotherapy—especially if recent—or external beam radiation therapy involving active bone marrow can worsen radionuclide-induced leukocytopenia and/or thrombocytopenia.
4.
Pregnancy must be excluded prior to radionuclide therapy, while breast feeding must be stopped.
5.
Current medications, especially those that can affect coagulation or blood cell counts, must be recorded.
6.
Estimation of life expectancy (life expectancy >3 months, Karnofsky index >70 %). A patient with a life expectancy of <3–4 weeks is unlikely to benefit from treatment. Similarly, patients showing rapidly progressing disease are not candidates for RIT because of delayed efficacy of the treatment.
3.2 Patient Information and Instruction
The treatment must be performed in close collaboration with the physicians treating the patient for the underlying disease.
Although most information will be collected by the haematologist, the NM physician should provide patients with guidelines on radiation therapy and written information describing the treatment, anticipated adverse events and contact telephone numbers.
Social interaction with relatives, friends and pets is without risk; however, there are some precautions that should be taken for 1 week following treatment with 90Y-ibritumomab-tiuxetan (see Fig. 1: an example of recommendation that we give our patients).
In accordance with other anticancer therapies, contraception should be used to avoid pregnancy for 1 year following treatment.
Fertility was considered unlikely to be affected in females, but temporary infertility is possible in males, with a low risk of permanent sterility, as 90Y-ibritumomab-tiuxetan treatment results in a radiation dose of 2.8 mGy/MBq to the testes (Schering; data on file (Wiseman et al. 2003b)). Male patients should therefore consider semen cryopreservation if prior therapies have not damaged sperm quality.
The patient should be advised of the risk of secondary malignancies, particularly myelodysplastic syndromes (MDS) and acute myeloid leukaemia (AML). The incidence of MDS and AML is low (1.4 %) following treatment with 90Y-ibritumomab-tiuxetan (Witzig et al. 2003) This incidence is within the range (1–8 %) previously reported with alkylating-based chemotherapy (Kantarjian and Keating 1987; Kyle et al. 2000; Pedersen-Bjergaard et al. 1985), and it is postulated that secondary malignancies after RIT may be attributable to exposure to alkylating agents during earlier treatments. To date, no secondary malignancies have been reported with the first-line use of RIT (Kaminski et al. 2003).
3.3 ASC Collection and Basal Evaluation
The ASC are collected via aphaeresis after mobilisation with growth factors (granulocyte colony-stimulating factor: GCSF) alone or together with chemotherapy (cyclophosphamide 2 g/m2), having a target of at least 2.0 × 109 CD34+/kg.
Mobilisation can be avoided in case of available PBSC from previous apheresis.
After collecting the CD34+, it is mandatory to perform a basal evaluation of the disease. Radiological-computed tomography scan (CT) of the neck, chest, abdomen and pelvis, CT/PET with 18F-FDG, bone marrow (BM) evaluation must be performed within 4 weeks prior to treatment. A cardiological and pulmonary evaluation at baseline and then at 4 weeks, 6 months and 1 year after 90Y-ibritumomab-tiuxetan administration are usually performed.
3.4 Dosimetry
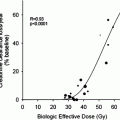
The first dosimetric data on patients treated with 90Yibritumomab-tiuxetan were obtained from multicenter trials for Food and Drug Administration approval (Wiseman et al. 2000; Zevalin 2007; US Food and Drug Administration 2007). It has been found that the spleen and the liver are the main source organs, red marrow is the critical organ and activity retention in the body is persistent. No correlation was observed between red marrow dose and toxicity effects, although the activity of 14.8 MBq/kg has been proved to limit haematologic toxicity.
As red marrow is thought to be the only limiting organ, dosimetry is not required for 90Y-ibritumomab-tiuxetan at the conventional activity of 14.8 MBq/kg (0.4 mCi/kg).
Although 90Y-ibritumomab-tiuxetan administered at high activity levels is likely to be myeloablative, it is not possible to identify either a “safe” maximal activity to be administered or a unique critical organ for all patients. The target limit of 20 Gy for the irradiation of the liver, in our high dose Zevalin® protocol, was considered an internal conservative choice in order to prevent unexplored risks (Ferrucci et al. 2007). Higher tolerance doses have been shown in external beam radiotherapy (EBR) (TD5 = 5¼23 Gy, kidneys; TD5 = 5¼30 Gy, liver) (Emami et al. 1991). There is also evidence that the tissue tolerance during radionuclide therapy may be higher than in EBR due to the difference in dose rates.
Nevertheless, the irradiation of vital organs in patients undergoing high-activity 90Y-ibritumomab-tiuxetan deserves special caution because of the clinical and medical history of these heavily pretreated patients: once combined with irradiation, the aftermath of toxicity from chemotherapy can generate serious and unpredictable effects.
In a paper we recently published (Aricò et al. 2009) about findings of abnormal dosimetry in scheduled high dose Zevalin® patients, we concluded that the clinical judgment of risk benefit in those patients derived from the dosimetric data: dosimetry is recommended when a patient is recruited in high activity Zevalin® protocols, in order to avoid usefulness and unjustified treatments, or serious toxic adverse events.
Before 111In-Ibritumomab-tiuxetan administration, patients receive a transmission scan using a 57Co-flood source, for possible attenuation correction (Savi et al. 2004), and a low-dose CT scan (80 mA), for evaluation of individual organ masses.
Then the patients receive the first infusion of rituximab (250 mg/m2), followed by 185 MBq of 111In-Ibritumomab-tiuxetan.
Following administration of 111In-Ibritumomab-tiuxetan, serial anterior/posterior whole-body scans are acquired at 0, 1, 16, 24, 96 and 144 h post to evaluate the biokinetics. Energy windows were set at the two 111In peaks (173 and 247 keV, 20 %) and at a third interval (140 keV, 9 %) for possible scatter correction (Cremonesi et al. 2002).
Blood and urine samples are also collected according to the same time schedule to determine the individual clearance of antibody from the blood.
Data are analysed according to the MIRD method, considering the conjugate view technique, in order to obtain predicted absorbed doses to non disease—involved organs (Cremonesi et al. 2007).
The absorbed doses are calculated using OLINDA/EXM software (Stabin et al. 2005), including individual organ masses. A conservative choice of 20 Gy to organs except red marrow can be defined as safe limit.
Related posts:
 (Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
 Thyroid Carcinoma
Thyroid Carcinoma
 of Progressive Dedifferentiated and Medullary Thyroid Cancer with Radiolabeled Somatostatin Analogs
of Progressive Dedifferentiated and Medullary Thyroid Cancer with Radiolabeled Somatostatin Analogs
 Bisphosphonates for Bone Pain Palliation
Bisphosphonates for Bone Pain Palliation
 Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
 Use of Dosimetry in the Planning of Patient Therapy
Use of Dosimetry in the Planning of Patient Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


