Chapter 75 Overview: Hypoplastic left heart syndrome (HLHS) is a spectrum of disease characterized by underdevelopment of the left ventricle with obstruction or atresia of ventricular inflow and outflow. HLHS accounts for approximately 2% to 3% of all cases of congenital heart disease, with a slight male predominance.1,2 Chromosomal abnormalities associated with HLHS include Turner syndrome, trisomy 13 and 18, and terminal 11q deletion.1–3 Etiology and Pathophysiology: The structural defects of HLHS include varying degrees of hypoplasia of left heart structures including a hypoplastic ascending aorta and arch, aortic valve atresia or stenosis, hypoplastic left ventricle, mitral atresia or stenosis, and patent ductus arteriosus (PDA) and patent foramen ovale or atrial septal defect (ASD) (Fig. 75-1). In a majority of cases, the ventricular septum is intact, and in severe cases, thickening of the left ventricular endocardium or endocardial fibroelastosis is present.4 Coarctation of the aorta coexists in approximately 80% of patients.5 The right heart structures often are enlarged.5 Figure 75-1 Diagram of hypoplastic left heart syndrome. Clinical Presentation: Cyanosis and tachypnea generally are apparent within hours to 2 days after birth. Poor arteriovenous mixing as a result of inadequate interatrial communication can lead to early elevation of left atrial and pulmonary venous pressure, pulmonary edema, and right heart failure. Serious hemodynamic changes occur after the birth of an infant with HLHS when pulmonary vascular resistance begins to drop and the PDA begins to spontaneously close.6 When pulmonary vascular resistance drops, an increase in pulmonary blood flow and a decrease in systemic blood flow occur, leading to a decrease in systemic perfusion. When the PDA constricts, a further decrease in the ductal-dependent systemic and coronary circulations occurs, leading to a decrease in systemic perfusion, myocardial ischemia, shock, and death. Imaging: HLHS is readily identified in utero. The condition is now discovered in approximately 60% of patients with use of prenatal echocardiography.7,8 Chest radiographic findings are variable. The heart may be normal in size or may be enlarged with a globular configuration, suggesting multichamber enlargement (e-Fig. 75-2). Pulmonary vascularity may be normal in the first hours of life, with a subsequent progressive increase in vascularity if no restriction to blood flow exists at the atrial level. Indistinctness of the pulmonary vessels or pulmonary venous congestion with interstitial lines or pleural fluid may be seen with a restrictive atrial septum. e-Figure 75-2 A newborn with hypoplastic left heart syndrome. Echocardiography is the imaging method of choice and generally delineates all relevant presurgical anatomy. Cross-sectional imaging can be performed as an adjunct to echocardiography in complex cases.9 Magnetic resonance imaging (MRI) may be especially useful when a marginally hypoplastic left ventricle is present and the possibility exists of a two-ventricle surgical repair (Fig. 75-3 and Video 75-1).4 In these cases, MRI can be used to assess the size of the left atrium, atrial septum, mitral valve orifice, left ventricle, and aortic root to help plan the most appropriate surgical repair. MRI increasingly is being used after the first stage of the palliative single ventricle surgery to reliably quantify the systemic right ventricle systolic function, tricuspid regurgitation, and residual or recurrent coarctation and for pulmonary artery stenosis or hypoplasia before the second-stage bidirectional cavopulmonary anastomosis.10–12 Figure 75-3 Hypoplastic left heart syndrome. Treatment: Initial medical management includes intravenous prostaglandin E1 (PGE1) to maintain ductal patency. Medical therapy, including ventilator adjustments, inhaled agents, and medications, is used to optimize the ratio of pulmonary to systemic blood flow and to minimize the volume load on the single functional ventricle to maintain adequate systemic perfusion.13 Surgical treatment consists of a staged reconstruction procedure (Fig. 75-4). The staged reconstruction involves three surgical procedures with the goal of creating separate pulmonary and systemic circulations supported by the right ventricle and accounts for the high neonatal pulmonary vascular resistance and the subsequent decrease in pulmonary vascular resistance. The first stage of the reconstruction—the Norwood procedure—often is performed in the first week of life.14 This procedure involves transection of the pulmonary trunk proximal to the pulmonary bifurcation and anastomosis of the pulmonary trunk to the aorta. A triangular patch of homograft material is used to augment the hypoplastic ascending aorta, aortic arch, and distal arch. The coronary arteries are then perfused retrograde through the small ascending aorta. The PDA is ligated. A complete atrial septectomy is performed. Blood flow to the lungs is reestablished via a modified Blalock-Taussig shunt (BT shunt) from the subclavian or brachiocephalic artery to the pulmonary artery. (Fig. 75-5). Alternatively, a right ventricle to pulmonary artery (RV-PA) conduit may be placed.15 The potential benefit of the RV-PA conduit is elimination of the diastolic runoff that occurs with a BT shunt. Diastolic runoff can lead to coronary and systemic artery blood flow steal or an increase in the ratio of pulmonary blood flow to systemic blood flow. The potential disadvantages of the RV-PA conduit include right ventricular dysfunction due to the ventriculotomy, the potential for aneurysm formation at the site of the conduit insertion, and right ventricular volume overload from pulmonary regurgitation because there is no valve in the conduit.1,15 Figure 75-4 Staged reconstruction for hypoplastic left heart syndrome. Figure 75-5 A 6-month-old with hypoplastic left heart syndrome after the Norwood I procedure. A “hybrid” approach, which combines surgical branch pulmonary artery banding to limit pulmonary blood flow with transcatheter ductal stenting to provide systemic flow, has more recently been advocated to achieve similar physiology to the stage I procedure without requiring cardiopulmonary bypass in the fragile neonate with HLHS (Fig. 75-6).1,16 The comprehensive second-stage procedure then involves cardiopulmonary bypass, removal of the PDA stent and pulmonary artery bands, repair of the aortic arch and pulmonary arteries, division of the diminutive ascending aorta with reimplantation into the pulmonary root, main pulmonary artery to reconstructed aortic anastomosis, atrial septectomy, and a bidirectional Glenn or hemi-Fontan procedure.1,16 Figure 75-6 A 5-month-old with hypoplastic left heart syndrome after a hybrid procedure. The third stage of the reconstruction—the Fontan completion procedure—is generally performed at 18 to 36 months of age. This stage involves directing inferior vena cava blood flow to the pulmonary arteries either via an extracardiac conduit or an intracardiac right atrial baffle (lateral tunnel). If a patch was placed in the right atrium during the second stage of the repair, it is removed. This third stage achieves separation of the systemic and pulmonary circulations. A fenestration may be left in the Fontan circuit so if pressures become high, there can be a pop-off from the Fontan circuit into the heart, which may lead to a more stable postoperative course.17,18 Many fenestrations close spontaneously but also can be closed during a cardiac catheterization procedure. Survival after the three-stage palliative procedure has improved steadily and now approaches 70%.8 The stage I Norwood procedure carries the highest mortality, ranging from 7% to 19%.15 Cardiac transplantation had been considered an alternative to staged Norwood palliation in the past, but because of limited donor availability and the recent improvement in survival following Norwood palliation, transplantation now generally is reserved for patients for whom staged palliation has failed.1,19 Overview: Left ventricular outflow tract (LVOT) obstruction can occur at the level of the aortic valve or in the subvalvar or supravalvar regions. This spectrum of disease represents approximately 10% of cases of congenital heart disease.20 Valvar aortic stenosis (AS) is by far the most common form of LVOT obstruction and has a male predominance of nearly 80%.21 Supravalvar AS accounts for 1% to 2% of AS in childhood and occurs spontaneously or may be familial, usually via autosomal dominant transmission. Up to 50% of patients with supravalvar AS have Williams syndrome. Subvalvar AS is slightly more common than supravalvar AS22; it also has a male predominance and may be associated with more complex disease such as double-outlet right ventricle and transposition of the great arteries. Etiology and Pathophysiology: Congenital aortic valve stenosis results from abnormal valve development rather than the degenerative disease commonly seen in adults. The stenotic valve has variable anatomy, with annular hypoplasia, thickened or tethered leaflets, and/or incompletely developed commissures. Valve morphology may predict clinical severity or associated disease.23,24 Whereas neonatal critical AS often is associated with a unicuspid valve and a small eccentric orifice, bicuspid morphology accounts for up to 95% of cases of congenital valvar AS and is present in up to 30% to 60% of patients with coarctation.25,26 In addition to the abnormality of the bicuspid valve, the aortic root tissue is abnormal in these patients and can lead to significant aortic dilation above a stenotic bicuspid valve. The aortic dilation has been thought to be due to poststenotic dilation, but histologic abnormalities of the ascending aorta can occur without significant valvar stenosis or regurgitation. The histology is similar to the medial disease seen in persons with Marfan syndrome in addition to abnormalities of the smooth muscle, extracellular matrix, elastin, and collagen.27,28 The narrowing in supravalvar AS most commonly is hourglass in shape, occurs immediately above the sinuses of Valsalva at the sinotubular junction, and may be associated with poststenotic dilatation of the aorta, diffuse aortic arch hypoplasia, aortic valve abnormalities, coronary artery ostial stenosis, or left ventricular hypertrophy.29,30 Supravalvar AS often is part of a widespread arteriopathy. Subvalvar AS can be discrete or diffuse. The discrete form, which represents the majority of cases, is a thin fibromuscular diaphragm encircling the LVOT. In the more severe diffuse form, a fibromuscular subaortic band is present along the length of the LVOT, producing a tunnel-like narrowing. The fibrous process often extends to involve the aortic valve cusps or the anterior mitral valve leaflet. The more diffuse form usually is associated with other left heart lesions, including mitral stenosis, supramitral ring, parachute mitral valve, valvar AS, or coarctation of the aorta.31 Clinical Presentation: Severe or critical AS may be diagnosed prenatally or present early in infancy with severe left heart obstruction, congestive heart failure, dyspnea, and poor peripheral circulation. Systemic flow is ductal dependent in cases of critical AS. The clinical manifestations of AS in infancy depend on the degree of valvar obstruction, mitral insufficiency, left atrial hypertension, and left ventricular dysfunction, as well as the amount of shunted atrial and ductal flow and other associated left-sided obstructive lesions. In cases of severe obstruction, the left ventricle may be severely hypoplastic, dilated, or dysfunctional. Imaging: The chest radiograph in infants with critical AS shows cardiomegaly and pulmonary venous congestion. In older children with mild stenosis, the radiograph generally is normal. When moderate to severe stenosis and left ventricular hypertrophy are present, the cardiac apex is depressed toward the diaphragm and posteriorly to the inferior vena cava. Left atrial enlargement can be seen with severe stenosis. Poststenotic dilation of the ascending aorta is rare in young children (Fig. 75-7). Figure 75-7 Aortic valve stenosis. Echocardiography is the imaging procedure of choice for the evaluation of valvar AS. Cardiac MRI can be used to complement echocardiography in cases of poor acoustic windows or if larger field of view imaging is indicated. Goals of imaging include demonstration of the degree and location of obstruction, valve morphology, leaflet mobility and effective valve orifice area. Systolic valve area calculated by MRI planimetry correlates well with transesophageal echocardiography and cardiac catheterization measurements (Fig. 75-8 and Video 75-2).32 The valve typically appears thickened and doming, with asymmetric or restricted leaflet excursion. The gradient across the aortic valve in millimeters of mercury is calculated using the modified Bernoulli equation (4V2, where V is peak Doppler velocity beyond the aortic valve in meters per second). Aortic valvular regurgitant fraction is calculated with phase-contrast MRI (e-Fig. 75-9). Evaluation of left ventricular size, systolic performance, and diastolic dysfunction is necessary. Assessment of aortic root, ascending aorta, and arch size is crucial in infants with critical AS. These patients may have abnormal endocardium, which can indicate the presence of endocardial fibroelastosis (e-Fig. 75-10). Surveillance of the thoracic aorta is indicated to evaluate for the development of aortic root dilation and aortic dissection associated with aortic valve disease (e-Fig. 75-11 and Video 75-3).28 Figure 75-8 Bicuspid aortic valve in a 25-year-old with history of aortic coarctation. e-Figure 75-9 Aortic valve flow curve.
Left Heart Lesions
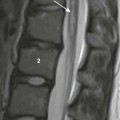
Hypoplastic Left Heart Syndrome
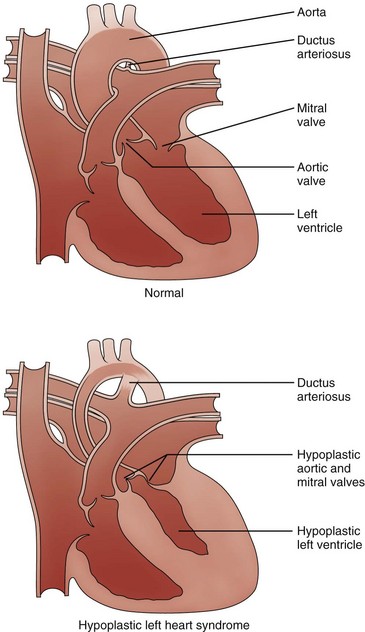
Compared with the normal heart, the mitral and aortic valves are severely hypoplastic or atretic, the left ventricular cavity and aorta are hypoplastic, and systemic blood flow is supplied by the patent ductus arteriosus. (From American Heart Association. Hypoplastic left heart syndrome. www.americanheart.org. © 2006, American Heart Association, Inc.)
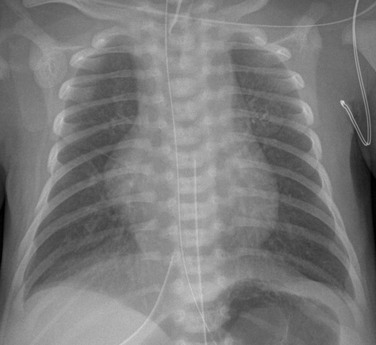
A frontal view of the chest shows a globular cardiac silhouette, consistent with multichamber enlargement. Mild perihilar haziness also is present, indicative of mild venous congestion.
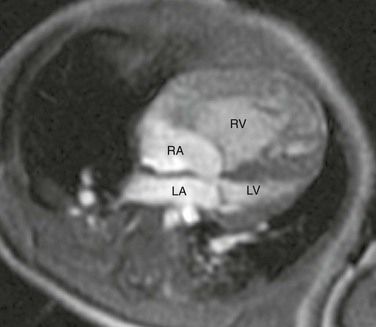
Four-chamber steady-state free-precession (SSFP) magnetic resonance image (MRI) of a 12-week-old with a marginally hypoplastic left ventricle to evaluate for a two-ventricle versus single-ventricle repair. The left atrium (LA) and left ventricle (LV) are small, with the left ventricular volume calculated at 24.2 mL/m2. The study also showed adequate mitral and aortic valve dimensions. Based on the MRI measurements, the patient underwent a successful two-ventricle repair. See Video 75-1 for a four-chamber cine SSFP MRI of the same patient. RA, Right atrium; RV, right ventricle.
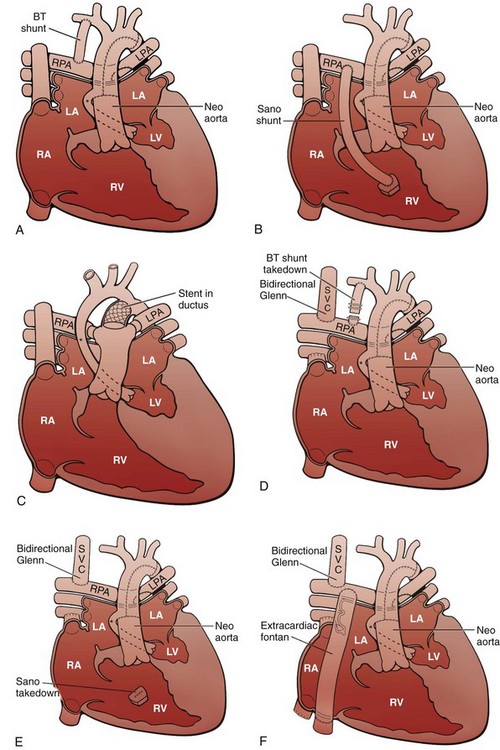
A, Stage I of the Norwood reconstruction using a modified Blalock-Taussig (BT) shunt. B, Stage I of the Norwood reconstruction using the Sano modification. C, Hybrid procedure. D, Stage II of the Norwood reconstruction. E, Stage II of the Norwood reconstruction using the Sano modification. F, Fontan procedure. Asterisk, Native ascending aorta; LA, left atrium; LPA, left pulmonary artery; LV, left ventricle; RA, right atrium; RPA, right pulmonary artery; RV, right ventricle; SVC, superior vena cava.
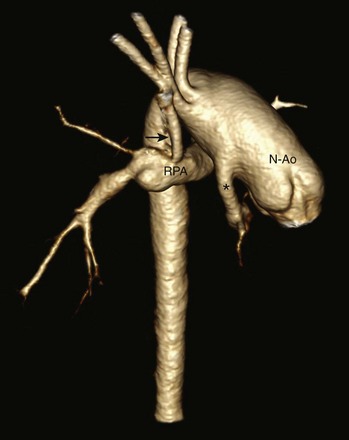
Posterior oblique volume-rendered computed tomographic angiography shows the hypoplastic native aorta (asterisk) anastomosed with the native pulmonary artery (now neoaorta) and a Blalock-Taussig shunt (arrow) extending from the right brachiocephalic artery to the right pulmonary artery. N-Ao, Neo-aorta; RPA, right pulmonary artery.
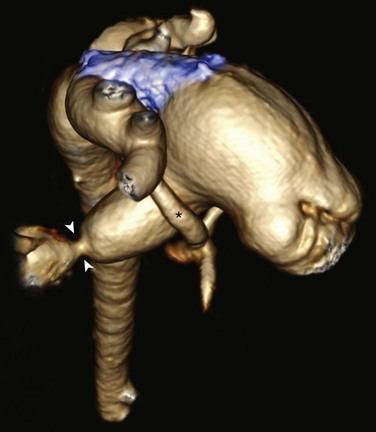
Posterior oblique volume-rendered computed tomographic angiography shows the hypoplastic native aorta (asterisk). A stent (blue) has been placed to keep the large patent ductus arteriosus open and a pulmonary artery band (arrowheads) has been placed to limit pulmonary blood flow.
Aortic Stenosis
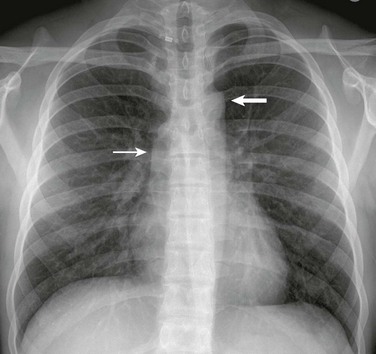
A frontal view of the chest in an 18-year-old shows poststenotic dilation of the ascending aorta (thin arrow) and aortic arch (thick arrow).
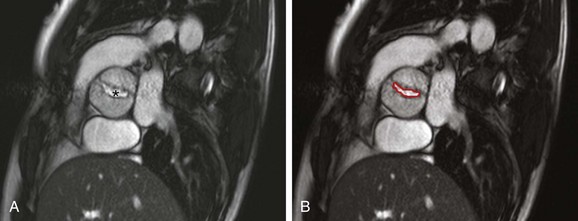
A, A cross-sectional steady-state free-precession (SSFP) systolic image of a severely stenotic bicuspid aortic valve (asterisk) with fusion of the right and noncoronary commissures, giving a “fish-mouth” appearance. See Video 75-2 for a cine SSFP image of the same. B, The aortic valve area (outlined in red) as measured by planimetry is 1.18 cm2.
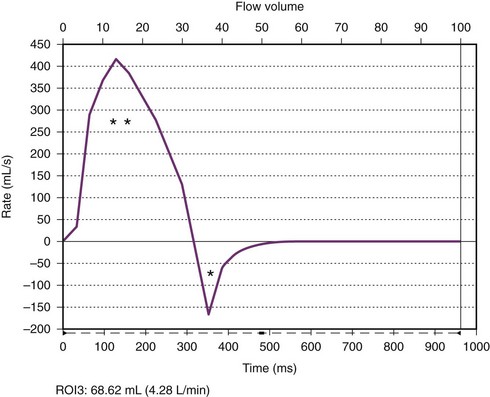
A flow curve obtained by magnetic resonance phase-contrast imaging acquired just above the aortic valve shows forward (two asterisks) and regurgitant (single asteriskRelated posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree