Disorders more prevalent in infants and young children
Diffuse developmental disorders
Acinar dysplasia
Congenital alveolar dysplasia
Alveolar capillary dysplasia with misalignment of the pulmonary veins
Growth abnormalities
Pulmonary hypoplasia
Chronic lung disease of infancy
Related to genetic disorders
Related to congenital heart disease
Specific disorders of unknown etiology
Pulmonary interstitial glycogenosis
Neuroendocrine cell hyperplasia of infancy
Surfactant dysfunction disorders
Related to a genetic defect
Related to autoimmune anti-GM-CSF antibody
Consistent histology without a recognized genetic defect
Disorders related to systemic disease processes
Autoimmune and autoinflammatory disorders
Vasculitis
Connective tissue disease
Immunodeficiencies
Primary
Secondary (drug therapy, radiation therapy, bone marrow transplant)
Lymphoid hyperplasia and lymphoproliferative disorders
Granulomatous disorders
Lysosomal storage disorders
Langerhans cell histiocytosis
Disorders of the normal immunocompetent host
Infectious and post-infectious processes
Bronchiolitis obliterans
Eosinophilic pneumonia
Related to exposures
Hypersensitivity pneumonitis
Aspiration pneumonia
Drug reaction
Toxic inhalation
Acute interstitial pneumonia
Cryptogenic organizing pneumonia
Idiopathic nonspecific interstitial pneumonia
Idiopathic pulmonary hemosiderosis
Vascular disorders masquerading as diffuse lung disease
Pulmonary venous congestion or obstruction
Congestive heart failure
Congenital cardiovascular disease
Pulmonary veno-occlusive disease
Lymphangiectasia
Lymphangiomatosis
The imaging technique and diagnostic efficacy of imaging for pediatric DLD have been addressed in previous review articles (Guillerman 2010; Guillerman and Brody 2011; Lee 2013). For some types of pediatric DLD, the imaging findings are highly specific, while for others the imaging findings are nonspecific and laboratory tests or lung biopsy are needed for definitive diagnosis (Kurland et al. 2013). In addition to suggesting or corroborating a specific diagnosis in some cases, imaging can be useful for refining the differential diagnosis, identifying biopsy sites, monitoring disease activity, and assessing response to therapy. The remainder of this chapter will provide an updated summary of the characteristic clinical and imaging features of the disorders categorized under a modified chILD classification scheme of pediatric DLD.
3 Categories of Disorders
3.1 Diffuse Developmental Disorders
Acinar dysplasia is characterized by lung developmental arrest in the pseudoglandular or early canalicular phase, resulting in essentially no alveolar spaces for gas exchange. Congenital alveolar dysplasia is characterized by arrest in the late canalicular/early saccular phase, resulting in incomplete alveolarization. Alveolar capillary dysplasia with misalignment of the pulmonary veins (ACD/MPV) is characterized by malpositioning of the pulmonary vein branches adjacent to the pulmonary artery branches rather than within the interlobular septa, medial hypertrophy of the pulmonary arterioles, reduced alveolar capillary density, and pulmonary lobular maldevelopment. Pulmonary lymphangiectasia is also present in about one-third of cases (Dishop 2011). The misaligned pulmonary veins in ACD/MPV may actually represent anastomotic bronchial veins associated with intrapulmonary right-to-left shunting. This shunting bypasses the alveolar capillary bed and exacerbates the hypoxemia from right-to-left extrapulmonary shunting associated with persistent pulmonary hypertension of the newborn (PPHN) (Galambos et al. 2014).
The diffuse developmental disorders of the lung are associated with markedly impaired alveolar gas exchange, resulting in respiratory failure and severe PPHN within hours or days of birth in the absence of the usual causative conditions of prematurity, meconium aspiration, congenital heart disease, perinatal asphyxia, or sepsis. Death usually ensues within a few days or weeks, unless the lung involvement is patchy rather than diffuse (Chow et al. 2013), or extracorporeal membrane oxygenation (ECMO) or paracorporeal lung assist devices are available as a bridge to lung transplantation (Sen et al. 2004; Michalsky et al. 2005; Hoganson et al. 2014). Approximately 80 % of cases of ACD/MPV are associated with extrapulmonary malformations, including hypoplastic left heart syndrome, aortic coarctation, alimentary tract atresia, annular pancreas, omphalocele, midgut malrotation, and urinary tract malformation. Sporadic or familial autosomal dominant FOXF1 gene mutations have been identified as a cause of some cases of ACD/MPV (Stankiewicz et al. 2009; Sen et al. 2013).
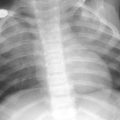
Due the severity of the respiratory disease, imaging of patients with diffuse developmental disorders of the lung is usually limited to portable CXRs. Although the initial CXRs may be unimpressive, followup CXRs may demonstrate progressive hazy opacification of the lungs, resembling of the findings of surfactant deficiency of prematurity or genetic surfactant disorders (Fig. 1). Pneumothorax or pneumomediastinum is common and likely attributable to barotrauma (Michalsky et al. 2005; Hugosson et al. 2005; Gillespie et al. 2004; Newman and Yunis 1990).
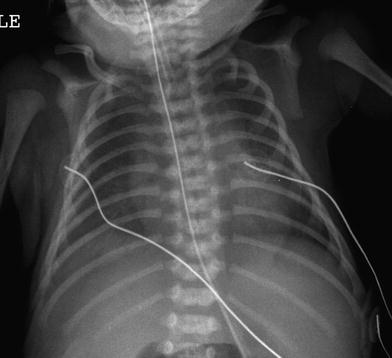
Fig. 1
Alveolar capillary dysplasia with misalignment of the pulmonary veins (ACD/MPV). A frontal portable CXR of a 33-week gestational age newborn with neonatal RDS shows diffuse, hazy pulmonary opacification indistinguishable from surfactant deficiency of prematurity. The patient rapidly developed hypoxemia and severe PPHN requiring ECMO
3.2 Growth Abnormalities
Growth abnormalities constitute the most common cause of chronic DLD in infancy. Lung growth abnormalities are characterized histologically by impaired alveolarization manifesting as lobular simplification with deficient alveolar vascularization and septation, reduced alveolar number, and increased alveolar size resembling emphysema (Dishop 2010). As a consequence, there is diminished total alveolar surface area and reduced pulmonary diffusing capacity relative to alveolar volume (Balinotti et al. 2010). Lung growth abnormalities are often accompanied by patchy PIG or by pulmonary arterial hypertensive changes related to increased vascular resistance from a deficient capillary bed (Dishop 2011).
Lung growth abnormalities can be related to prenatal or postnatal conditions (Deutsch et al. 2007). The most common prenatal form is pulmonary hypoplasia secondary to limited intrathoracic space in utero, such as from a congenital diaphragmatic hernia, oligohydramnios, thoracic mass lesion, or skeletal dysplasia. The most frequently encountered postnatal form is chronic lung disease of infancy (CLDI) related to prematurity. This includes “new” BPD, which is characterized by impaired alveolarization, but less fibrosis and airway obstruction compared to classic BPD (Bhandari and Bhandari 2009), and Wilson-Mikity syndrome, which is characterized by slowly progressive respiratory distress and cyst-like changes of the lungs developing within the first few weeks of life despite minimal respiratory support at birth (Hoepker et al. 2008; Philip 2009). Growth abnormalities can also be observed in near-term and term infants as an idiopathic disorder or in association with congenital heart disease or certain genetic disorders (Deutsch et al. 2007; Dishop 2010).
Patients with a lung growth abnormality typically present with respiratory difficulty as a neonate, and may either improve or worsen, depending on the extent of catch-up growth of the alveoli over time and whether or not pulmonary hypertension ensues. Most alveolarization occurs by two to three years postnatal age, but recent evidence from hyperpolarized gas magnetic resonance imaging suggests that neo-alveolarization continues to occur throughout childhood and adolescence in normal individuals (Narayanan et al. 2012). Gestational age at birth is an important determinant of subsequent alveolar development (Balinotti et al. 2010), but the capacity for catch-up neo-alveolarization in those with lung growth abnormalities is currently unknown.
The chest imaging findings of infants with BPD and other lung growth abnormalities range from nearly normal to markedly abnormal, with distorted pulmonary lobules of variable shape and attenuation, perilobular reticular opacities, linear subpleural opacities, ground-glass opacities, and foci of decreased lung attenuation, often appearing as cyst-like hyperlucencies (Metwalli et al. 2011) (Fig. 2). The decreased lung attenuation is likely attributable to enlarged alveoli with reduced alveolar septation and vascularization (Mahut et al. 2007). Decreased lung attenuation on CT correlates with the clinical severity of BPD (la Tour et al. 2013). Reproducible quantitative CT scoring systems based on the extent of findings, such as decreased lung attenuation, linear opacities, and architectural distortion at a mean postmenstrual age of 39 weeks show better correlation than do CXR scores with the clinical severity of BPD, and higher CT scores predict a higher risk of rehospitalization for pneumonia (Shin et al. 2013). CT scoring systems are superior to pulmonary function testing for differentiating infants with and without a history of BPD and higher CT scores are associated with a greater number of days on supplemental oxygen (Sarria et al. 2011). Ventilation-perfusion (V/Q) single photon emission computed tomography (SPECT) performed at a mean postmenstrual age of 37 weeks commonly reveals V/Q mismatching, even in clinically mild BPD, and the degree of V/Q mismatching correlates with the duration of mechanical ventilation (Kjellberg et al. 2013). Residual pulmonary abnormalities on CT are very common in adolescent and young adult survivors of classic BPD born in the pre-surfactant era, with the most frequent findings being subpleural, triangular, and linear opacities, air trapping, and emphysema (Aukland et al. 2006; Wong et al. 2008, 2011) (Fig. 3). The imaging findings in adolescent and adult survivors of “new” BPD are not yet well described. The imaging findings of BPD are further discussed in the chapter entitled Neonatal Chest Imaging by Crotty in this book.
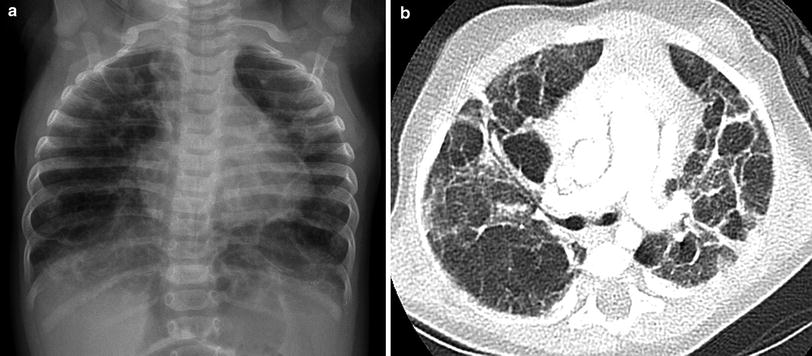
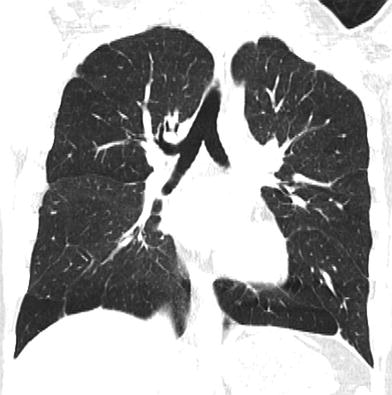
Fig. 2
Growth abnormality. A frontal CXR (a) of a five-month-old, former 32-week preemie with CLDI and continued supplemental oxygen requirement shows pulmonary hyperinflation, reticular opacities, and cystic changes. An axial chest CT image (b) demonstrates architectural distortion with ground-glass opacities, thick perilobular opacities, and variably-sized pulmonary lobules, some of which exhibit cyst-like hyperlucency related to alveolar simplification
Fig. 3
Growth abnormality. A coronal chest CT image from a 15-year-old former preemie with a history of BPD depicts multiple subpleural triangular and linear opacities, and hyperlucent areas resembling emphysema. These findings are commonly observed in adolescent survivors of BPD
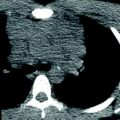
Down syndrome is associated with a peculiar lung growth abnormality in which the enlargement of the alveoli and alveolar ducts is particularly marked in the subpleural region, resulting in the appearance of subpleural cysts on CT (Biko et al. 2008) (Fig. 4). A lung growth abnormality should be suspected in children with Down syndrome and respiratory difficulties not attributable to congenital heart disease.
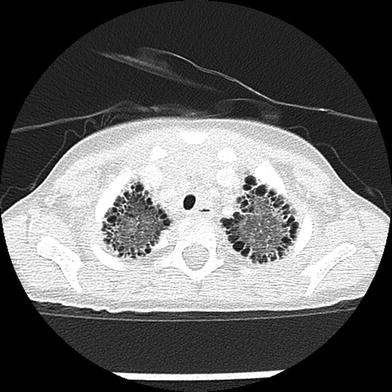
Fig. 4
Growth abnormality. An axial chest CT image from a two-year-old with Down syndrome shows numerous bilateral subpleural cysts. Enlargement of the alveoli and alveolar ducts is particularly marked in the subpleural region in the lung growth abnormality related to Down syndrome
A severe progressive lung growth disorder ultimately requiring lung transplantation for survival can develop in infants with X-linked filamin A (FLNA) gene mutations. Chest imaging in these patients characteristically shows severe pulmonary hyperinflation and hyperlucency of multiple lobes of the lungs resembling emphysema along with atelectasis and airway malacia. Extrapulmonary manifestations in patients with FLNA gene mutations include periventricular nodular gray matter heterotopia, nystagmus, joint hyperextensibility, congenital heart defects, and vascular aneurysms (Guillerman 2010; Masurel-Paulet et al. 2011; de Wit et al. 2011; Guillerman et al. 2013) (Fig. 5).
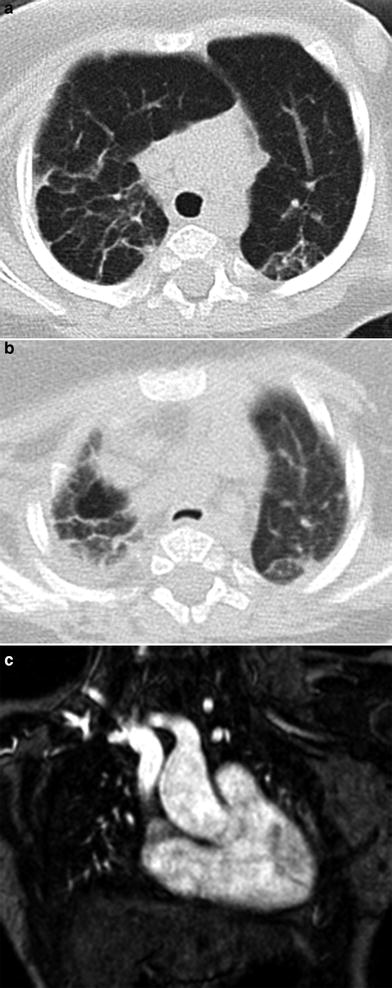
Fig. 5
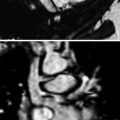
Growth abnormality. An axial chest CT inspiratory image (a) from a six-month-old girl with an X-linked filamin A gene mutation demonstrates severe bilateral pulmonary hyperinflation and scattered atelectasis. The corresponding axial chest CT expiratory image (b) reveals narrowing of the trachea related to tracheomalacia. A coronal chest MR angiography image (c) obtained at 2 years of age shows dilation of the ascending aorta and innominate artery
3.3 Specific Disorders of Unknown Etiology
3.3.1 Pulmonary Interstitial Glycogenosis
PIG, previously known as infantile cellular interstitial pneumonitis, is characterized histologically by patchy or diffuse infiltration of the interstitium by vimentin-positive immature mesenchymal cells containing abundant cytoplasmic glycogen, without prominent inflammation or fibrosis (Smets et al. 2004). PIG has not been observed beyond infancy, suggesting a relationship of PIG to lung development and growth (Deterding 2010; Deutsch and Young 2010). Patchy PIG is often observed concomitantly with a lung growth abnormality (Deutsch et al. 2007), and may contribute to some exacerbations of CLDI (Dishop 2011) or persistent pulmonary hypertension in infants with congenital heart disease (Radman et al. 2013).
Most patients with PIG present in early infancy with tachypnea and a supplemental oxygen requirement. Pulmonary function testing reveals a restrictive lung disease with marked reduction of pulmonary diffusing capacity. The observations that resolution of PIG on histology and improvement in pulmonary diffusion capacity and forced vital capacity coincide with clinical improvement suggest that PIG impairs respiratory function via alveolar septal thickening (Ehsan et al. 2014), although the ultimate clinical outcome primarily depends upon the severity of any underlying lung growth abnormality (King et al. 2011), and no mortality is associated with cases of isolated PIG. Corticosteroid therapy may hasten the resolution of PIG, possibly due to acceleration of lung maturation rather than to suppression of inflammation (Deterding 2010; Deutsch and Young 2010; Canakis et al. 2002; Onland et al. 2005), but should be used judiciously in this self-limited disorder due to the risks of neuro-developmental impairment, immunosuppression, and poor wound healing associated with corticosteroids (Radman et al. 2013).
Reported features of PIG on CXR include hyperinflation and interstitial opacities (Smets et al. 2004; Canakis et al. 2002; Onland et al. 2005; Lanfranchi et al. 2010). Reported features of PIG on CT include pulmonary architectural distortion, interstitial thickening, ground-glass opacities, and hyperlucent areas (Smets et al. 2004; Onland et al. 2005; Lanfranchi et al. 2010; Castillo et al. 2010) (Fig. 6). However, these reports describe findings that are at least in part attributable to coexisting lung growth abnormalities. The imaging appearance of PIG in the absence of a concomitant lung growth abnormality is unknown at present, and lung biopsy is currently required for diagnosis.
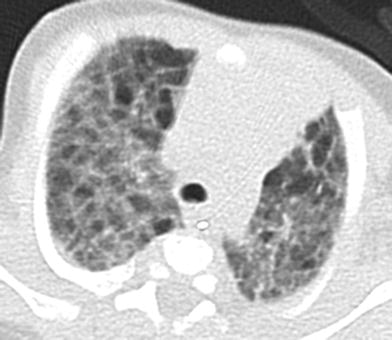
Fig. 6
Pulmonary interstitial glycogenosis. Axial chest CT image from a 7-week-old, former 29-week preemie with hypoxemia shows both ground-glass opacities and hyperlucent pulmonary lobules. Lung biopsy revealed a growth abnormality with superimposed alveolar wall thickening related to pulmonary interstitial glycogenosis
3.3.2 Neuroendocrine Cell Hyperplasia of Infancy
Neuroendocrine cell hyperplasia of infancy (NEHI), also known as persistent tachypnea of infancy, is characterized histologically by increased numbers of pulmonary neuroendocrine cells (PNECs) and innervated clusters of PNECs called neuroepithelial bodies in the epithelium of the peripheral airways (Deterding et al. 2005). Detection of PNECs on histology is facilitated by special staining for bombesin. PNECs are involved in oxygen sensing and fetal lung development, and rapidly decline in number in normal individuals after the neonatal period. The absence of other significant airway or interstitial disease is an important criterion for the diagnosis of NEHI, since increased numbers of PNECs can also be observed in dissimilar conditions, such as sudden infant death syndrome, BPD, pulmonary hypertension, cystic fibrosis, mechanical ventilation, and smoke exposure (Dishop 2011). NEHI may be associated with minor patchy inflammation or fibrosis in a small proportion of airways (Young et al. 2010). The existence of familial cases of NEHI suggests a genetic etiology for some cases (Popler et al. 2010). Heterozygous mutations in the NK2 homeobox 1 (NKX2-1) gene have been reported in a NEHI case, but are not suspected as the predominant cause of NEHI (Young et al. 2013).
NEHI usually presents by six months of age in full-term infants with persistent tachypnea, hypoxemia, and slow weight gain. There is a male predominance. Auscultation may reveal crackles, but wheezing is unusual. The anteroposterior diameter of the chest is often increased. Symptoms can be precipitated or exacerbated by viral respiratory infections or residence at high altitude (Gomes et al. 2013). Lung function tests show peripheral airway obstruction and profound air trapping with reduced forced expiratory flow (FEV) and markedly elevated functional residual capacity (FRC) and residual volume (RV) (Kerby et al. 2013; Lukkarinen et al. 2013). The severity of air trapping as measured by FRC and RV inversely correlates with room air oxygen saturation at short-term (6–12 months) followup, providing a possible prognostic marker. Compared to BPD patients, NEHI patients have similar degrees of airway obstruction, but greater air trapping (Kerby et al. 2013). The severity of airway obstruction correlates with the prominence of PNECs (Young et al. 2010). Treatment is largely supportive and focused on oxygen supplementation and nutritional support. Bronchodilators and corticosteroids have not been shown to be beneficial except for treatment of superimposed viral infections (Kerby et al. 2013; Lukkarinen et al. 2013). NEHI is not a life-threatening condition and most patients show clinical and radiographic improvement, especially after two years of age. However, some patients require supplemental oxygen for months to years, and air trapping and exercise intolerance may persist into adolescence (Deterding 2010; Gomes et al. 2013). The development of nonatopic asthma has also been reported in follow-up (Lukkarinen et al. 2013).
On CXR, NEHI manifests with hyperinflation similar to bronchiolitis or reactive airways disease, but without airway wall thickening. CT findings of diffuse air trapping with mosaic attenuation and geographic ground-glass opacities of the right middle lobe, lingula, parahilar, and paramediastinal lung regions are highly characteristic of NEHI, approaching 100 % specificity in the appropriate clinical setting (Fig. 7). Ground-glass opacities may also be present at the peripheral posterior aspects of the lungs. Consolidation, nodules, septal thickening, cysts, and bronchiectasis are usually absent, and the presence of these findings suggests a superimposed or alternative disorder. NEHI is unable to be excluded by CT, since the sensitivity of CT for NEHI is approximately 80 %, and missed diagnosis may occur when the ground-glass opacities are faint or distributed in an atypical pattern (Brody et al. 2010). Due to variability in the number of PNECs, pathologic confirmation of the diagnosis is not always reliable, especially if airway sampling is limited in the biopsy specimen (Young et al. 2010). A presumptive diagnosis of NEHI can be made without lung biopsy if the clinical presentation and the findings on CT or pulmonary function testing are characteristic (Deterding 2010; Young et al. 2010; Brody et al. 2010; Kerby et al. 2013; Gomes et al. 2013).
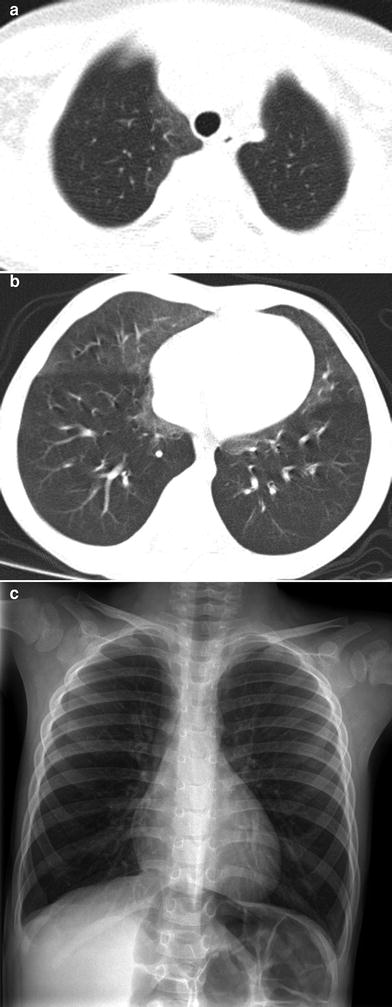
Fig. 7
Neuroendocrine cell hyperplasia of infancy. Axial chest CT images (a, b) from a 26-month-old with persistent tachypnea show pulmonary hyperinflation and ground-glass opacities in the right middle lobe, lingula, and paramediastinal regions in a distribution characteristic of neuroendocrine cell hyperplasia of infancy. A frontal CXR (c) obtained at 3 years of age shows continued pulmonary hyperinflation
3.4 Surfactant Dysfunction Disorders
3.4.1 Genetic Disorders of Surfactant Metabolism
Pulmonary surfactant is composed primarily of phospholipids and surfactant proteins secreted by type II alveolar cells. The lowering of intra-alveolar surface tension by surfactant is required for normal respiratory function. Surfactant also plays a role in innate host defense. Surfactant is cleared by uptake into alveolar epithelial cells or alveolar macrophages under stimulation by granulocyte-macrophage colony-stimulating factor (GM-CSF) (Suzuki et al. 2010).
Genetic disorders of surfactant metabolism are a primary cause of unexplained fatal respiratory distress syndrome (RDS) in term neonates, and are increasingly identified as a cause of chronic DLD in older infants, children, and adolescents. The most frequently identified disease-causing mutations involve the genes encoding ATP binding cassette A3 (ABCA3) (Hamvas 2010) and surfactant protein C (SP–C) (Nathan et al. 2012). Other disease-causing mutations may involve the genes encoding surfactant protein B (SP–B), colony-stimulating factor 2 receptor alpha (CSFRA), NK2 homeobox 1 (NKX2-1), and solute carrier family 7 amino acid transporter member 7 (SLC7A7) (Nogee 2010).
In young infants presenting with a genetic disorder of surfactant metabolism, histologic findings usually consist of PAP with diffuse alveolar epithelial hyperplasia and foamy macrophages without hyaline membrane formation. With aging, there tends to be a lesser degree of PAP, and histologic findings of lobular remodeling, cholesterol clefts, variable interstitial fibrosis, and interstitial inflammation develop in a CPI or DIP pattern later in infancy or childhood, or in a NSIP pattern later in childhood or adolescence. Endogenous lipoid pneumonia may also be observed. These overlapping histologic patterns limit genotypic-phenotypic correlation, although electron microscopy can be useful for identifying abnormal lamellar bodies that are characteristic of ABCA3 mutations (Dishop 2010).
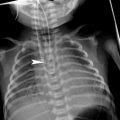
The typical clinical presentations and imaging findings of the surfactant dysfunction disorders vary with patient’s age. Term neonates unable to produce SP–B due to disease-causing biallelic inherited loss-of-function autosomal recessive SP–B gene mutations develop severe RDS within hours of birth. CXRs show diffuse hazy granular pulmonary opacification similar to RDS of prematurity (Fig. 8). However, in contrast to RDS of prematurity, infants with genetic SP–B deficiency progress to respiratory failure that is not ameliorated by exogenous surfactant administration. In the absence of lung transplantation, most die within a few months after birth. In rare cases with mutations that allow some SP–B production, longer term survival may be feasible (Nogee 2010; Clement et al. 2010).
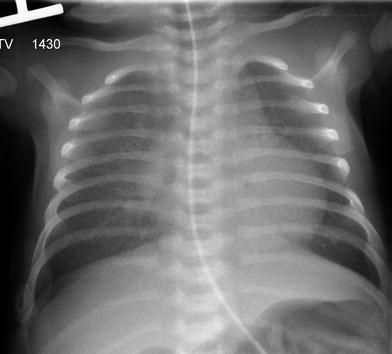
Fig. 8
Surfactant metabolism disorder related to SP–B gene mutation. A frontal CXR of a 12-day-old, full-term infant with respiratory distress demonstrates diffuse hazy granular pulmonary opacities resembling the findings usually associated with surfactant deficiency of prematurity
Variable phenotypes ranging from acute severe RDS in term neonates to chronic DLD in children and adolescents are associated with spontaneous or inherited autosomal dominant SP–C gene mutations (Guillot et al. 2009; Thouvenin et al. 2010) or autosomal recessive ABCA3 gene mutations (Doan et al. 2008; Flamein et al. 2012). A common clinical presentation of DLD related to SP–C or ABCA3 gene mutations is cough, tachypnea, and hypoxemia beginning in infancy. Alternatively, affected individuals can remain asymptomatic for years despite progressive fibrosis before sudden life-threatening deterioration occurs. Heterozygosity for an ABCA3 mutation can modify the severity of DLD associated with an SP–C mutation (Bullard and Nogee 2007). Interestingly, monoallelic ABCA3 mutation carriers are overrepresented in infants of >34 weeks gestation age with RDS (Wambach et al. 2012). Treatment with corticosteroids, hydroxychloroquine, or azithromycin may be associated with clinical improvement of patients with genetic disorders of surfactant metabolism, but it is uncertain whether this is entirely due to the therapy or in part to the natural history of the disease, and there are no proven curative therapies except for lung transplantation in the terminal disease phase (Deterding 2010; Nogee 2010; Thouvenin et al. 2010).
CXRs of young infants with disease-causing SP–C or ABCA3 gene mutations demonstrate diffuse or patchy hazy granular pulmonary opacities, while CT scans show diffuse ground-glass opacities, consolidation, septal thickening, or crazy-paving (Doan et al. 2008; Olsen et al. 2004; Stevens et al. 2005; Prestridge et al. 2006; Soraisham et al. 2006). In older infants and children, the ground-glass opacities tend to decrease and thin-walled cysts develop and increase in number and size with age (Doan et al. 2008; Mechri et al. 2010; Flamein et al. 2012) (Fig. 9). Possibly as an effect of chronic restrictive lung disease on the growing chest wall, pectus excavatum often develops in patients with DLD related to ABCA3 mutations who survive infancy (Doan et al. 2008). Changes in lung findings on CT do not correlate with lung function or outcome in patients with DLD related to ABCA3 mutations, and there is no established role for routine chest imaging after the diagnosis has been established (Doan et al. 2008).
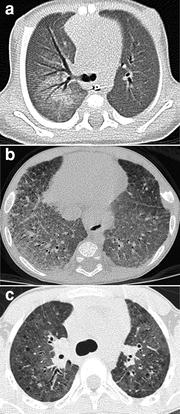
Fig. 9
Surfactant metabolism disorder related to SP–C gene mutation. An axial chest CT image (a) from a 5-week-old shows diffuse ground-glass opacities and right parahilar consolidation. An axial chest CT image (b) from a five-month-old depicts diffuse ground-glass opacities and septal thickening. An axial chest CT image (c) from a 9-year-old reveals patchy ground-glass opacities and scattered small parenchymal cysts
Understanding of the spectrum of genetic disorders associated with surfactant dysfunction and pediatric DLD continues to broaden. NK2 homeobox 1 (NKX2-1), also known as thyroid transcription factor-1 (TTF-1), is expressed in the forebrain, thyroid, and lung, and regulates transcription of the genes for surfactant proteins and Clara cell secretory protein. Disease-causing sporadic or inherited autosomal dominant NKX2-1 gene mutations or deletions can lead to maldevelopment of the basal ganglia and thyroid and decreased production of surfactant proteins, resulting in “brain-lung-thyroid syndrome” characterized by congenital hypothyroidism, hypotonia, chorea, and lung disease. Only a slight majority of cases manifest with the full syndrome, and there is isolated lung disease in one-fifth of cases at presentation. Mortality related to the lung disease is around 20 % (Carre et al. 2009; Hamvas et al. 2013). Clinical manifestations of the lung disease are varied and illustrate the diverse role NKX2-1 plays in surfactant function, alveolarization, and innate immunity. The presenting pulmonary phenotype is severe neonatal RDS in about three-fourths of cases and chronic childhood DLD in about one-fifth of cases. Recurrent respiratory infections are also common (Guillot et al. 2010; Hamvas et al. 2013). The histology is heterogeneous, and may include changes of PAP, CPI, DIP, NSIP, or growth abnormality with alveolar simplification. CT findings include diffuse or patchy ground-glass opacities, consolidations, and cysts (Galambos et al. 2010; Hamvas et al. 2013) (Fig. 10). Thyroid ultrasonography or scintigraphy reveals thyroid hypoplasia, hemiagenesis or athyreosis in 45 % of cases (Carre et al. 2009). Intriguingly, a case of NEHI associated with NKX2-1 mutations has been reported, suggesting that NEHI may result from altered expression of genes regulated by NKX2-1 other than those in the surfactant system (Young et al. 2013).
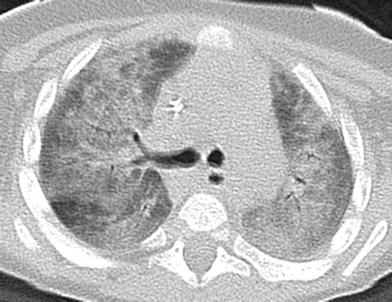
Fig. 10
Surfactant metabolism disorder related to NKX2-1 gene mutation. Bilateral patchy ground-glass opacities are shown on an axial chest CT image from a 13-month-old with chorea, hypotonia, hypothyroidism, and chronic pneumonitis of infancy as manifestations of the “brain-lung-thyroid” syndrome
Homozygous or compound heterozygous mutations or deletions of the gene encoding colony-stimulating factor 2 receptor alpha (CSF2RA), also known as granulocyte-macrophage colony-stimulating factor receptor alpha (GM-CSFRA), can severely reduce GM-CSF receptor signaling and impair alveolar macrophage function. This results in PAP that is characterized by filling of alveoli with surfactant and foamy macrophages and preservation of normal alveolar walls, in contrast to the disorders of surfactant production associated with SP–B, SP–C, or ABCA3 mutations, in which alveolar epithelial hyperplasia and lobular remodeling occur. Children with heredity PAP related to CSF2RA gene mutations or deletions develop progressive dyspnea, exercise intolerance, or tachypnea at 1–9 years of age, with a mean age of 5 years at symptom onset. The clinical course is often insidious and initial misdiagnosis as asthma or pneumonia is common. Serum and bronchoalveolar (BAL) fluid characteristically show elevated GM-CSF levels. The PAP is typically patchy, leading to patchy pulmonary airspace opacities on CXR and geographic ground opacities and crazy-paving on chest CT (Suzuki et al. 2008; Martinez-Moczygemba et al. 2008; Suzuki et al. 2010). Whole-lung lavage therapy results in clinical and radiographic improvement (Fig. 11), in contrast to patients with disorders of surfactant production who do not respond well to lavage therapy (Suzuki et al. 2010).
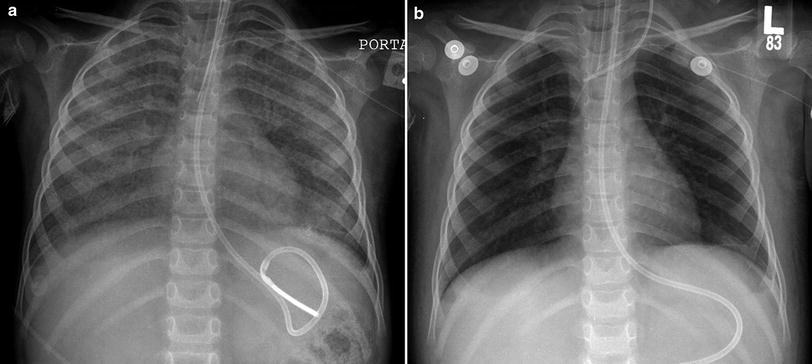
Fig. 11
Surfactant metabolism disorder related to CSF2RA mutation. A frontal CXR (a) of a 3-year-old demonstrates diffuse pulmonary airspace opacification. Disrupted GM-CSF receptor signaling in this genetic disorder results in impaired alveolar macrophage function and pulmonary alveolar proteinosis. Dramatic improvement in lung aeration is shown on a frontal CXR (b) following whole-lung lavage
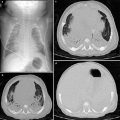
Lysinuric protein intolerance (LPI) is a multisystem disease resulting from an inherited defect of cationic amino acid transport attributable to autosomal recessive SLC7A7 gene mutations. LPI is associated with dietary protein intolerance, failure to thrive, osteoporosis, hepatosplenomegaly, hemophagocytic lymphohistiocytosis, immune dysfunction, nephropathy, and lung involvement (Ogier de Baulny et al. 2012). The lung involvement can occur at any age, including childhood, and is characterized by endogenous lipoid pneumonia, PAP, or pulmonary hemorrhage. CT is a sensitive test for diagnosing early lung involvement that manifests as septal thickening, nodules, and subpleural cysts prior to the onset of respiratory symptoms or pulmonary function test abnormalities (Santamaria et al. 1996). Some patients rapidly progress to respiratory failure with widespread pulmonary airspace opacities on CXR and CT related to PAP from alveolar macrophage dysfunction (Parto et al. 1993; Ogier de Baulny et al. 2012). LPI is generally treated with dietary protein restriction and L-citrulline supplementation. The lung involvement can be treated with corticosteroids and whole-lung lavage (Ogier de Baulny et al. 2012).
Recognition of clinical and radiographic findings suggestive of a surfactant dysfunction disorder is important, since the diagnosis may then be confirmed by genetic testing for surfactant gene mutations, avoiding the risk of lung biopsy. Lung biopsy may still be appropriate if genetic testing is nondiagnostic or if awaiting genetic testing results would delay the diagnosis in patients with rapidly progressive disease requiring lung transplantation for continued survival (Doan et al. 2008; Mechri et al. 2010). Occasionally, cases are encountered with imaging and histologic findings suggestive of a surfactant dysfunction disorder, but genetic testing for known disease-causing mutations is negative. Some of these cases are likely related to as yet characterized genetic defects of surfactant metabolism, especially in those with a family history of unexplained childhood-onset DLD.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


