Fig. 1
Patent ductus arteriosus in a 3-month-old girl with pulmonary hypertension. a Chest X-ray shows right pulmonary hyperinflation and a bulge in the left upper cardiac silhouette. b CT demonstrates that it corresponds to a huge patent ductus. c The right main pulmonary bronchus is compressed by the right pulmonary artery causing obstructive emphysema of the right lung
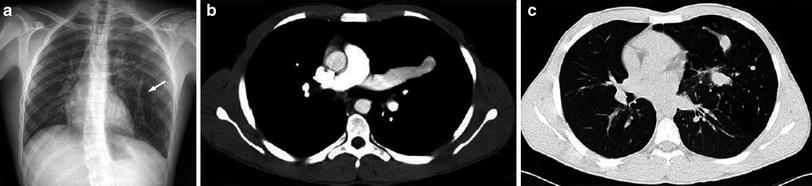
Fig. 2
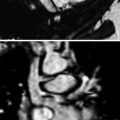
17-year-old boy with chest pain. a Chest radiograph shows a hyperinflation of the lingula and a vascular linear shadow (arrow) b, c CT demonstrates that this structure corresponds to a huge single pulmonary vein going to the left atrium. Hyperinsuflation of the lingula is also seen
2.1.2 Bronchial Atresia
Bronchial atresia (BA) is an anomaly characterized by obliteration of the proximal lumen of a segmental bronchus, with preservation of the distal structures. Its pathogenesis is unknown, although it may be due to a vascular insult. Most pediatric BA cases are congenital, but the condition may be acquired secondary to traumatic or infectious injury to the bronchus (Wasilewska et al. 2012). Air enters the affected segment via collateral channels, producing overinflation and air-trapping. The mucus secretions generated in the bronchi accumulate at the point of obstruction, originating mucus impaction (Lemire et al. 1970; Felson 1979). The mucocele can be linear, branched, ovoid, or spherical. BA almost always affects just one segment, and rarely affects a lobar bronchus. Involvement of multiple segments has been reported in a few cases (Ward and Morcos 1999). BA is characteristically located in the left upper lobe (apico-posterior segment), but can involve any lobe (Remy-Jardin et al. 1989; Medelli et al. 1979) and can be associated with other congenital anomalies. It is usually asymptomatic and is an incidental finding on radiological study. Infection of the unconnected lung is rare.
The chest plain film usually demonstrates pulmonary insufflation with trapped air during expiration, accompanied by a tubular, branched, or spherical image in a central position, which corresponds to the mucocele (Jederlinic et al. 1986). CT shows the segmental overinflation and mucous impaction with great precision. When BA does not involve the left upper lobe or when it does not present characteristic radiological findings in the plain film, CT is diagnostic, demonstrating the combination of emphysema and bronchial impaction that is the hallmark of this condition (Pugatch and Gale 1983; Finck and Milne 1988) (Fig. 3). In some cases, a cystic lesion containing gas and fluid corresponding to a severely dilated bronchus just distal to segmental bronchial atresia can also be seen (Griscom 1993) (Fig. 4).
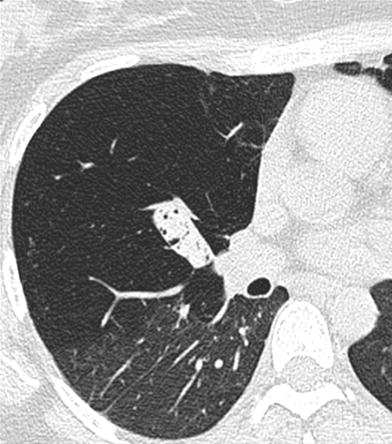
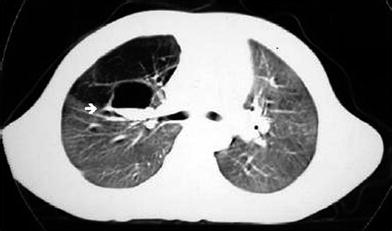
Fig. 3
Bronchial atresia in a 17-year-old girl. Expiratory CT image with lung window shows the bronchocele and hyperinflation of the right upper lobe
Fig. 4
Bronchial atresia in a 4-year-old boy. CT scan imaged with lung window shows a cystic lesion containing gas and fluid at the right hilum (arrow) corresponding to the dilated right upper lobe bronchus and hyperinflated right upper lobe
When the mucocele cannot be identified, radiological diagnosis of bronchial atresia versus CLE may be impossible. In both these entities symptoms may be absent and radiological features may progressively improve. Nonetheless, the degree of mediastinal shift and collapse of the ipsilateral lobes is much more significant in most cases of lobar emphysema than in bronchial atresia (Castellote et al. 2005).
MRI is not commonly used to evaluate bronchial atresia after birth, but this malformation can be depicted on prenatal MRI. The obstructed lobe appears hyperintense on T2-weighted images. This appearance can also be seen in CLE, type III CPAM, and BPS (Recio Rodríguez et al. 2012; Biyyam et al. 2010).
2.2 Single Congenital Thoracic Cyst
We include under the term single congenital thoracic cyst (SCTC) all congenital cysts located in the mediastinum (bronchogenic cysts, duplication cysts, and pleuropericardial cysts) and lung parenchyma. Treatment of SCTC depends on the symptoms. The best approach in asymptomatic patients with mediastinal cysts is periodic control, avoiding surgery. For practical purposes of clinical management, all cysts located within the lung parenchyma can be considered bronchogenic cysts requiring surgery. The definitive diagnosis of SCTC should be established on the basis of the study of the cyst wall. When there is associated inflammation and in some cases of mediastinal cysts, diagnosis can be difficult.
The most frequent location of bronchogenic cyst varies according to the published series (DuMontier et al. 1985; Baker 1989; Patcher and Lattes 1963; Rogers and Osmer 1964). In the most recent series including 68 bronchogenic cysts (McAdams et al. 2000), 58 (85 %) were mediastinal, and seven were intrapulmonary (10 %), demonstrating a clear predominance of mediastinal cysts. Mediastinal cysts are most often found in a subcarinal location, whereas intrapulmonary bronchogenic cysts are most frequently located in the lower lobes. Bronchogenic cysts can be found in the diaphragm, below the diaphragm (Braffman et al. 1988) and even in the liver (Kimura et al. 1990) or neck and can be associated with pericardial agenesis (Kwak et al. 1971). They are usually solitary and spherical in shape with thin walls of bronchial epithelium, and have a viscous gelatinous, mucoid, hemorrhagic or watery, translucent fluid content. They occasionally contain calcium, have calcified walls, or are completely calcified and they can be air-filled when they communicate with the bronchial tree (Rogers and Osmer 1964; Reed and Sobonya 1975). Bronchogenic cysts are sometimes found in association with other congenital pulmonary malformations such as sequestration, lobar emphysema, or BA (Kuhn and Khun 1992; Grewal and Yip 1994).
In infants mediastinal SCTC tend to compress or distort the esophagus, the trachea, and bronchi, resulting in clinical respiratory compromise, but the condition can be asymptomatic and be discovered fortuitously. Compression of a main bronchus may result in obstructive pulmonary hyperinflation of the ipsilateral lung (Fig. 5). These cysts can also compress the pulmonary artery or superior vena cava (Bankoff et al. 1985). Mediastinal and intrapulmonary SCTC can disappear spontaneously (Martin et al. 1988), change form due to decreases in their internal pressure, or diminish in size, making themselves invisible to the chest plain film (Fig. 6).
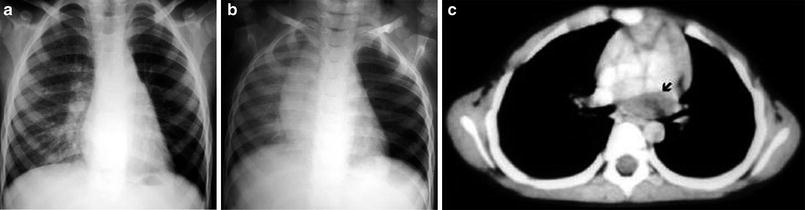
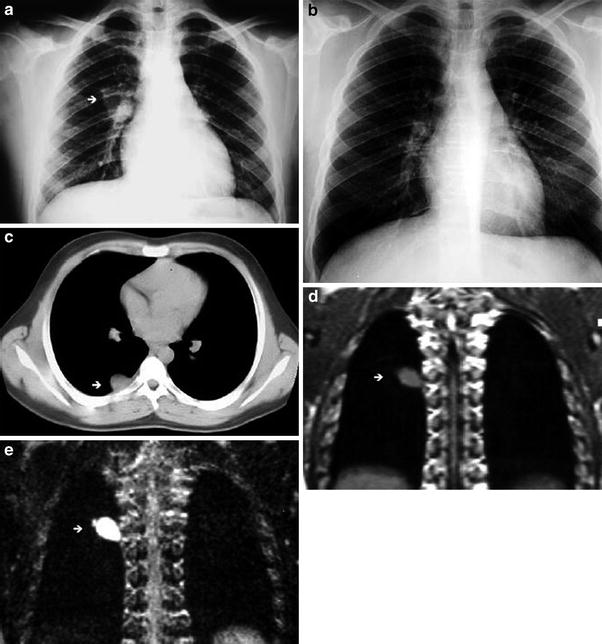
Fig. 5
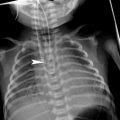
Bronchogenic cyst in a 3-year-old boy. a Inspiratory chest radiograph shows hyperlucency and decreased vascular perfusion in the left lung. b Expiratory chest radiograph demonstrates air-trapping in the left lung. c A subcarinal cyst compressing the left main bronchus is seen in the CT scan (arrow)
Fig. 6
Mediastinal bronchogenic cyst in a 14-year-old boy. a Chest radiograph shows an ovoid perihilar mass in the right upper lobe (arrow). b Chest radiograph performed 1 year later, prior to surgery, is normal. c CT scan performed at the same time as (b), shows an isodense mass in the right upper lobe (arrow). d Coronal T1-weighted MR image demonstrates the presence of an intermediate signal intensity ovoid mass (arrow). e Coronal T2-weighted MR image. The homogeneous high signal intensity of the mass indicates a fluid content (arrow)
The basic radiological study used to detect SCTC is the chest plain film. In the majority of cases, this technique detects the lesion and some of the complications (e.g., compression on neighboring structures). Ultrasound, MDCT, and MR allow a better evaluation of SCTC and its anatomic relationship with adjacent structures. CT reveals a round or ovoid mass with water or soft-tissue attenuation. Almost 50 % of SCTC appear iso- or even hyperdense at CT due to intracystic hemorrhage, protein content, or milk of calcium. On MRI, SCTC are homogeneously and markedly hyperintense on T2-weighted images. The intracystic signal intensity on T1-weighted images is more variable, and, depending upon the cyst content, low, intermediate, and high signal intensity cases have been reported (Naidich et al. 1988; Nakata et al. 1993). Relatively high signal intensity on T1 is due to a high protein content and or the presence of methemoglobin. Fluid–fluid levels have also been reported (Lyon and McAdams 1993). When air–fluid levels are seen within the cyst, it is usually infected, although we have seen cysts with air–fluid levels in asymptomatic patients (Fig. 7). Minimal wall enhancement is expected with gadolinium enhancement.
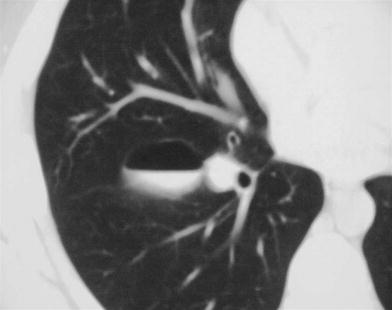
Fig. 7
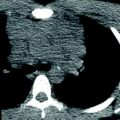
Bronchogenic cyst in an asymptomatic 12-year-old boy. CT scan shows an air–fluid level within the cyst
2.3 Congenital Pulmonary Airway Malformation: Pulmonary Sequestration Complex
2.3.1 Congenital Pulmonary Airway Malformation
Congenital pulmonary airway malformation, previously named congenital cystic adenomatoid malformation (CCAM) consists of an intrapulmonary mass of disorganized pulmonary tissue that may or may not be accompanied by macroscopic cysts (Stocker 2002). When present, the cysts communicate with the airways and their vascular supply comes from pulmonary circulation. However, there are numerous examples of CPAM fed by systemic blood vessels and in these cases it is extremely difficult to differentiate CPAM from pulmonary sequestration, as they correspond to overlapping malformations (Winters et al. 1997). From the radiological viewpoint the differentiation between CPAM with systemic supply and pulmonary sequestration is impossible. These malformations correspond to the same clinical and radiological entity, although they have a different anatomo-pathological expression.
Ch’in and Tang applied the name “congenital cystic adenomatoid malformation” to a congenital cystic pulmonary anomaly for the first time in 1949. The essential discovery is an adenomatoid proliferation of the terminal bronchioles that produces cysts of varying sizes coated with bronchial epithelium. There is considerably controversy over classification and nomenclature. CPAM has recently been recommended as a preferred term to congenital cystic adenomatoid malformation because not all the lesions are cystic and only type III is adenomatoid. In 1977 Stocker et al. divided CCAM into three groups, depending on whether the cysts were larger than 2 cm (type I), smaller than 2 cm (type II), or the malformation was solid without cysts (type III). (Stocker et al. 1977). An expanded classification (types 0–4) has been proposed representing malformations of larger through smaller airways. In an attempt to maintain a similar ordering of the three types published in 1977, type I, II, and III become type 1, 2, and 3 and the “distal acinar” lesion added is type 4, presenting features of a large cyst generally located at the periphery of the lung. Type 0 is characterized by its involvement of all lobes of the lung and its incompatibility with life (Table 1). A number of reports have been suggesting a relationship between CPAM and pleuropulmonary blastoma (PPB) and it is unclear at this time. Anyway, although Stocker feels based in his experience that they are separate lesions, other authors (MacSweeney et al. 2003) consider that both lesions show histologic overlap.
Table 1
The extended classification of congenital pulmonary airway malformation
Type | Incidence (%) | Gross appearance | Microscopy | Other features |
|---|---|---|---|---|
0 | 1–3 | Solid; both lungs are small and firm | Bronchial-type airways that have cartilage, smooth muscle, and glands are separated by abundant mesenchymal tissue | Incompatibility with life |
1 | 60–70 | Large cysts (up to 10 cm) | The cysts are lined by pseudostratified ciliated cells that are often interspersed with rows of mucous cells | May be late; best overall prognosis; less than 1 % carcinomatous change |
2 | 10–15 | Sponge-like composed of multiples cysts (up to 2 cm) and solid pale tumor-like tissue | The cysts resemble dilated bronchioles separated by normal alveoli; striated muscle in 5 % | Neonates; cardiac and renal anomalies; poor prognosis |
3 | 5 | Solid | Scattered bronchiolar/alveolar duct-like structures are lined by low cuboidal epithelium and surrounded by alveoli lined by cuboidal epithelium | Neonates, almost exclusively in males; poor prognosis |
4 | 15 | Large cysts (up to 10 cm) generally at the periphery of the lung | The cysts are lined by a flattened epithelium resting on loose mesenchymal tissue | Neonates and infants; good prognosis; overlap with PPB |
CPAM can be associated with other congenital malformations such as pulmonary sequestration, tracheal bronchus, tracheal atresia, tracheal diverticulum (Restrepo et al. 2004), and bronchogenic cyst. CPAM can also be associated with congenital BA in the same lobe (Cachia and Sobonya 1981) or can involve more than one lobe or be bilateral. In our experience it occurs with more frequency in lower lobes. CPAM can cause severe respiratory distress in the neonatal period. Beyond this time it is usually discovered when it becomes infected or as an incidental radiological finding (Pulpeiro et al. 1987).
The malformation can escape detection on plain films. Although, these days, however, CPAM is increasingly being diagnosed on antenatal (US) examinations, where it is seen as an echogenic mass, which may or may not contain cysts. Antenatal MR imaging may help to evaluate any associated pulmonary hypoplasia and predict the prognosis. The lesions may disappear completely, may remain unchanged or increase in size and be associated with the development of polyhydramnios or fetal nonimmune hydrops fetalis (Fig. 8) (Paterson 2005).
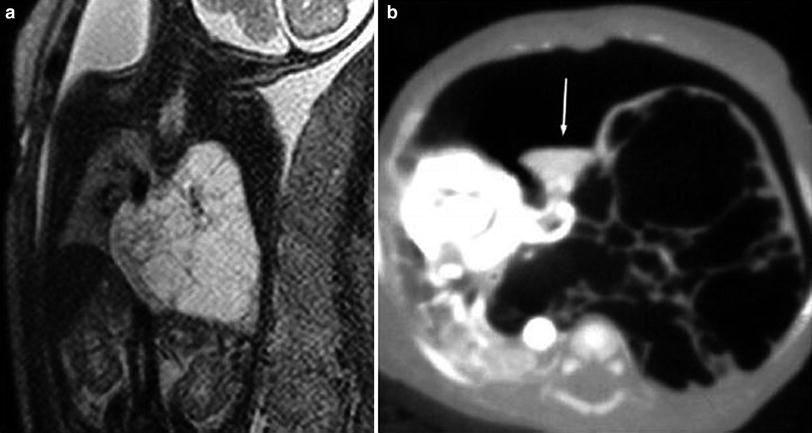
Fig. 8
CPAM type 1. a Coronal MR haste image in a 27-week-old fetus shows a large high signal intensity slightly heterogeneous mass with multiple septi arising from the left lung and crossing the midline. b A CT done when the patient was 5-day-old shows an anterior pneumothorax and multiple cysts occupying the left hemithorax, crossing the midline, displacing the heart to the right. A small collapsed superior lobe is seen (arrow)
The appearance of CPAM on radiographs and CT depends on the relative presence of cystic and solid components and whether there is superimposed infection. On plain films, type I presents as one or more dominant cysts with adjacent smaller cysts and solid tissue elements. Type II displays smaller, more evenly sized and spaced cysts. Type III, which is very rare, appears as a solid mass. Large masses produce significant mediastinal displacement. Air-fluid levels are often seen, mainly, but not always, associated with infection. On chest CT, CPAM is seen as multiple thin- or thick-walled, air- or fluid-filled cysts of variable size, expanding the affected lung. Air-fluid levels are often present (Rosado-de-Christenson and Stocker 1991). CPAM may mimic cystic pleuropulmonary blastoma. PPB is probably the same tumor that several authors have reported as mesenchymal sarcoma or rhabdomyosarcoma arising in congenital lung cysts (Ueda et al. 1977). It has also been stated that PPB can arise from preexisting cystic lung disease, but is reasonable to assume that the cystic changes are a component of the PPB, itself (Murphy et al. 1992). As the initial manifestation, pneumothorax is more frequently seen in PPB than in CPAM (Fig. 9) (Senac et al. 1991). However, pneumothorax can be found associated with both entities and the available information does not support the use of this finding as a differentiating diagnostic criteria (Lejeune et al. 1999).
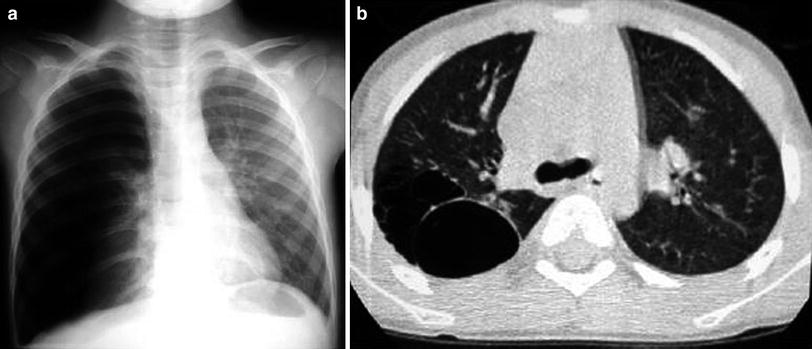
Fig. 9
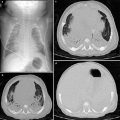
Congenital pulmonary airway malformation type I in a 6-year-old girl with chest pain and dyspnea. a Chest radiograph shows right-sided pneumothorax. A chest tube was inserted and the lung was re-expanded. b Chest CT performed 1 week later reveals multiple cysts at the right upper lobe
Cases of CPAM/pulmonary sequestration that dramatically decreased in size or disappear completely during pregnancy and infancy have been reported (Fig. 10). However, the clinical management of an asymptomatic child with a congenital mass of the lung remains controversial. Some authors advocate close clinical observation and radiological surveillance (MacGillivray et al. 1993), whereas others, considering the possibility that cystic pulmonary lesions may be infected (Garcia-Peña et al. 2013), or may be harbor or develop PPB, favor elective surgical resection (Samuel and Burge 1999).
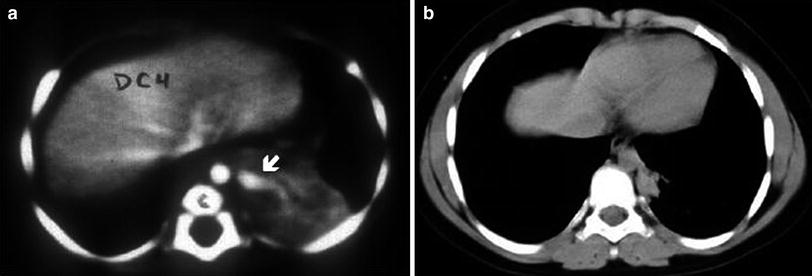
Fig. 10
Spontaneous involution of a pulmonary sequestration. a Contrast-enhanced chest CT at the age of 3 months shows a soft tissue density mass with a large feeding vessel originating from the aorta (arrow). b Significant shrinkage of the mass is seen in the scan at the age of 10 years
2.3.2 Pulmonary Sequestration
Pulmonary sequestration consists of a mass of pulmonary tissue disconnected from the bronchial tree that receives its blood supply from the systemic circulation (Heitzman 1984). Pulmonary sequestration is divided into two groups: intralobar sequestration, in which the tissue is surrounded by normal lung and found in the interior of the visceral pleura, and extralobar sequestration, in which the tissue is disconnected from the bronchial tree and has its own pleural coating. In 1946 Pryce identified intralobar sequestration as a clinical-pathological entity. He was the first to apply the term “sequestration” and further classified the lesion as intralobar or extralobar on the basis of the morphologic patterns of the malformation. There are also mixed cases with characteristics of both intralobar and extralobar sequestration.
Pulmonary sequestration is an uncommon anomaly; the intralobar form is more frequent than the extralobar. Intralobar sequestration constitutes 75 % of all pulmonary sequestrations and is located in the left lower lobe in 60 % of the cases. Only 2 % occur in the upper lobes and 0.25 % in the middle lobe. The affected lung can maintain the normal lung architecture, behave like a mass, or present as internal cysts. Vascularization in the majority of cases is through the thoracic aorta (Savic et al. 1979), or less commonly, through systemic vessels originating from the abdominal aorta or one of its branches (Pedersen et al. 1988). The systemic supply can be formed by multiple, small-caliber blood vessels or by a single vessel, which are histologically similar to the pulmonary artery. This favors the early appearance of atherosclerosis (Ikezoe et al. 1990). Intralobar sequestration does not receive blood from the pulmonary arteries and it drains through pulmonary veins. In exceptional cases it communicates with the digestive tract (bronchopulmonary foregut malformation) (Hruban et al. 1989). It can be bilateral, or associated with other congenital malformations.
Since Pryce’s description of pulmonary sequestration, there has been considerable controversy about its origin. Some authors contend that intralobar sequestration is, in fact, acquired (Gebauer and Mason 1959; Holder and Langston 1986), resulting from endobronchial obstruction leading to chronic pulmonary infection and hypertrophy of the systemic arteries in and around the area of the pulmonary ligament. This explains why, in the past, when infections were poorly controlled, intralobar sequestration was overdiagnosed. However, congenital intralobar sequestration does occur, since this anomaly has been recognized on prenatal US and has been detected in newborns (West et al. 1989; Laurin and Hägertrand 1999) (Fig. 11).
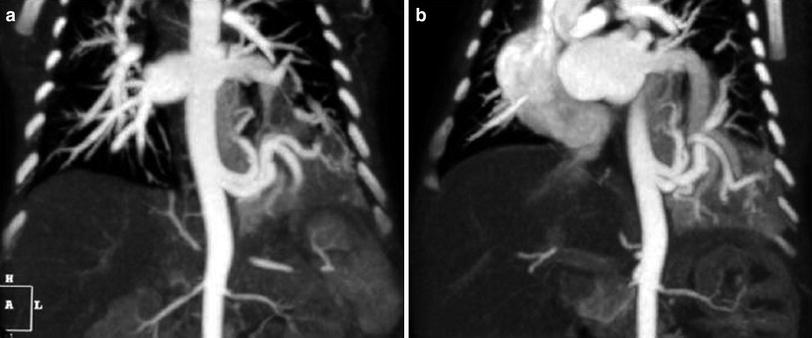
Fig. 11
Intralobar pulmonary sequestration in a 10-day-old boy with an echogenic mass in the left lower lobe seen on prenatal US. a Coronal MIP CT reconstructed image shows two anomalous arteries arising from thoracic aorta. b The drainage is through the lower left pulmonary vein
Intralobar sequestration is usually discovered because the patient has developed a pulmonary infection, although some patients are asymptomatic when the lesion is found. In a small number of cases intralobar sequestration debuts as a pulmonary hemorrhage (Fig. 12), pleural effusion (Kim et al. 1997; Lucaya et al. 1984), or pleural bleeding secondary to infarction of the sequestrated lung (Zumbro et al. 1974). On plain films, intralobar sequestration appears as a homogenous opacity mostly in the lower lobes. This opacity can simulate a mass with a well-defined border, or show internal air–fluid levels and a poorly defined border. In rare cases calcifications are present within the sequestration or in the systemic blood vessel. An unusual presentation of intralobar sequestration is localized emphysema without an associated opacity or mass (Ko et al. 2000).
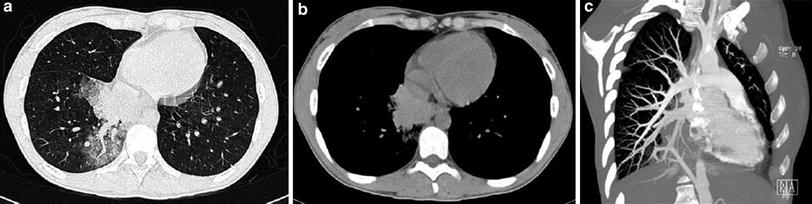
Fig. 12
Pulmonary sequestration in a 18-year-old with hemoptysis. a CT demonstrates the presence of ground glass area due to hemorrhage in the right lower lobe with a central vessel. b Shows a hyperdense mass secondary to hemorrhage into sequestration. c Angio-CT with MIP reconstructions shows that the vessel originates from the abdominal aorta
Extralobar sequestration is usually located between the lower lobe and the diaphragm, more frequently at the left thoracic base, in 77 % of cases (Savic et al. 1979). To a much lesser degree it has also been found in the mediastinum, pericardium (Stocker and Kagan-Hallet 1979), diaphragm, or retroperitoneum (Baker et al. 1982). Vascular supply occurs through a systemic artery and venous drainage through the azygos or portal system (Rees 1981), although nearly 25 % are completely or partially drained by pulmonary veins. Extralobar sequestration is associated with other congenital malformations in 65 % of cases, such as diaphragmatic hernia, bronchogenic cyst, BA, scimitar syndrome, pericardial defect and as previously stated, CCAM (Savic et al. 1979), and these are much more frequent than those associated with the intralobar form. Both types of sequestration occasionally can connect with the digestive tract (bronchopulmonary foregut malformation) (Hruban et al. 1989). The presence of air bronchograms within a mass thought to be ELS should suggest the diagnosis of bronchopulmonary foregut malformation. Communication between the tracheobronchial tree and the GI should be examined by upper GI series or MDCT. Extralobar sequestration is usually detected fortuitously and can also be associated with pleural effusion. It may be seen as a homogeneous mass or a small bump on the posterior hemidiaphragm that may be subtle and occasionally inapparent on the chest radiograph.
Ultrasound, CT, and MRI are useful in the study of sequestrations (Fig. 13). Multidetector CT angiography is considered the imaging technique of choice for preoperative evaluation of pulmonary sequestration. MDCT with supplementary multiplanar and 3D images has the advantage of being able to show the pulmonary parenchyma abnormality, as well as the arterial and venous angioarchitecture of the sequestration (Lee et al. 2004, 2011a). CT has some advantages over MR angiography for evaluating pulmonary sequestrations in children. CT scan times are significantly shorter, and resolution of small vessels and lung parenchyma is better. CT in a single phase of contrast injection is adequate for evaluation of both the arterial and the venous anatomy, using low dose to minimize radiation (Abbey et al. 2009; Lee et al. 2011b). Intralobar sequestration typically manifests as a homogeneous or inhomogeneous solid mass, with or without definable cystic changes. It can also appear as an aggregate of multiple small cystic lesions with air or fluid content (Fig. 14), a well-defined cystic mass, or a large cavitary lesion with air–fluid level. The lesion may enhance with contrast material (Frazier et al. 1997; Rosado-De-Christenson et al. 1993). An appearance simulating emphysema, possibly resulting from collateral ventilation and air-trapping, can sometimes be seen in sequestration. Expiratory CT scans are helpful for delineating the extent of the malformation (Fig. 15) (Stern et al. 2000; Lucaya et al. 2000). Extralobar sequestration is seen on chest CT as a homogeneous, well-delimited mass, sometimes with internal cystic areas (Rosado-De-Christenson et al. 1993).
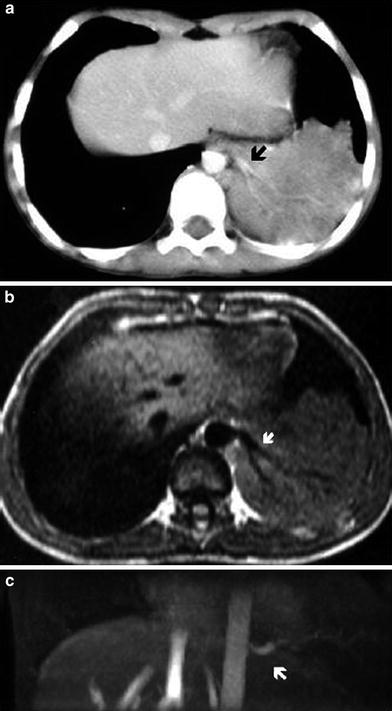
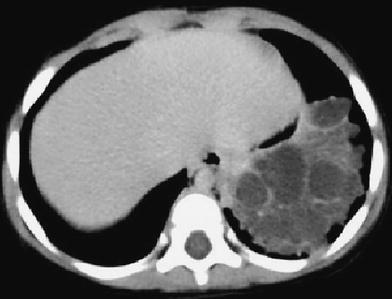
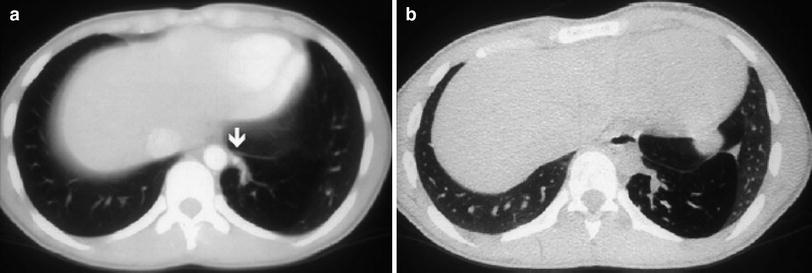
Fig. 13
Intralobar pulmonary sequestration of the left lower lobe in a 2-year-old boy. a Contrast-enhanced chest CT shows the sequestration and a systemic branching vessel originating from the aorta (arrow). b Axial SE T1-weighted and c coronal 2D TOF MIP reconstruction images demonstrate a branching feeding vessel originating from the descending aorta and supplying the pulmonary sequestration (arrows)
Fig. 14
Intralobar pulmonary sequestration in a 2-year-old boy. CT scan shows a mass with multiple fluid-filled cysts in the left lower lobe
Fig. 15
Intralobar pulmonary sequestration in left lower lobe in a 15-year-old boy. a Enhanced CT scan shows hyperlucency in left lower lobe. A systemic vessel (arrow) originating from the aorta and feeding the sequestration is well defined. b In an expiratory high-resolution CT scan at same level as (a), air-trapping can be seen within the sequestered lung. (Reprinted with permission)
MR imaging is a safe and noninvasive imaging method, which may be useful in some specific cases of pulmonary sequestration (Naidich et al. 1988; Yu et al. 2010). In MR, this anomaly is seen as a well-defined, irregular, or branch-like mass. MR can also reveal the presence of cystic areas, as well as the variable solid, fluid, hemorrhagic, and mucus-containing components. However, MR imaging cannot delineate focal thin-walled cysts or the emphysematous changes of sequestration as clearly as CT. The size, origin, and course of the aberrant systemic artery and the venous drainage can be demonstrated by MR imaging. Breath-hold (if possible) or nonbreath-hold three-dimensional contrast-enhanced MR angiography offer excellent display of the aberrant vessel without flow artifacts (Ko et al. 2000). The advantage of MRI over CT is the absence of radiation. However, long scan times, sedation requirements, and suboptimal evaluation of lung parenchyma are the disadvantages of this method.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


