1. Malformations secondary to abnormal neuronal and glial proliferation and apoptosis
A. Severe congenital microcephaly (MIC)
B. Megalencephaly (MEG)
C. Cortical dysgenesis with abnormal cell proliferation but without neoplasia
D. Cortical dysgenesis with abnormal cell proliferation and neoplasia
2. Malformations due to abnormal cortical migration
A. MCD with neuroependymal abnormalities: periventricular heterotopia
B. MCD due to generalized abnormal transmantle migration
C. MCD presumably due to localized abnormal late radial or tangential transmantle migration
D. MCD due to abnormal terminal migration and defects in pial limiting membrane
3. Malformations due to abnormal postmigrational development
A. MCD with polymicrogyria (PMG) or cortical malformations resembling PMG
B. Cortical dysgenesis secondary to inborn error of metabolism
C. Focal cortical dysplasia due to late developmental disturbances
D. Postmigrational developmental microcephaly
4.2 Malformations Secondary to Abnormal Neuronal and Glial Proliferation or Apoptosis
This group consists of three main entities: congenital microcephalies, megalencephalies, and focal/diffuse dysgenesis and dysplasia. The latter group is further subdivided in cortical dysgenesis with abnormal cell proliferation but without neoplasia and in cortical dysgenesis with abnormal cell proliferation and neoplasia.
4.2.1 Congenital Microcephaly
Microcephaly is usually defined as head circumferences of two standard deviations (SD) or more below the mean. When congenital microcephaly is the only abnormality found on evaluation, the disorder has been designated as primary microcephaly [6]. Most genes known to cause primary microcephaly affect pathways involving neurogenesis in particular the cell cycle phases of mitosis. From a diagnostic point of view, the microcephalic fetal brain is not only a “small brain” but shows some peculiar aspects: smooth and underdeveloped frontal and temporal opercula with a general lesser development of the frontal region (Fig. 4.1). These findings are quite evident after birth, the neonatal microcephalic brain presents with small frontal lobes and small frontal opercula, and also the frontal horns of lateral ventricle appear as the smaller part of the lateral ventricles (Fig. 4.2). In case of severe microcephaly, oligogyria ensues, and usually the more severe microcephaly is, the more severe the oligogyria.
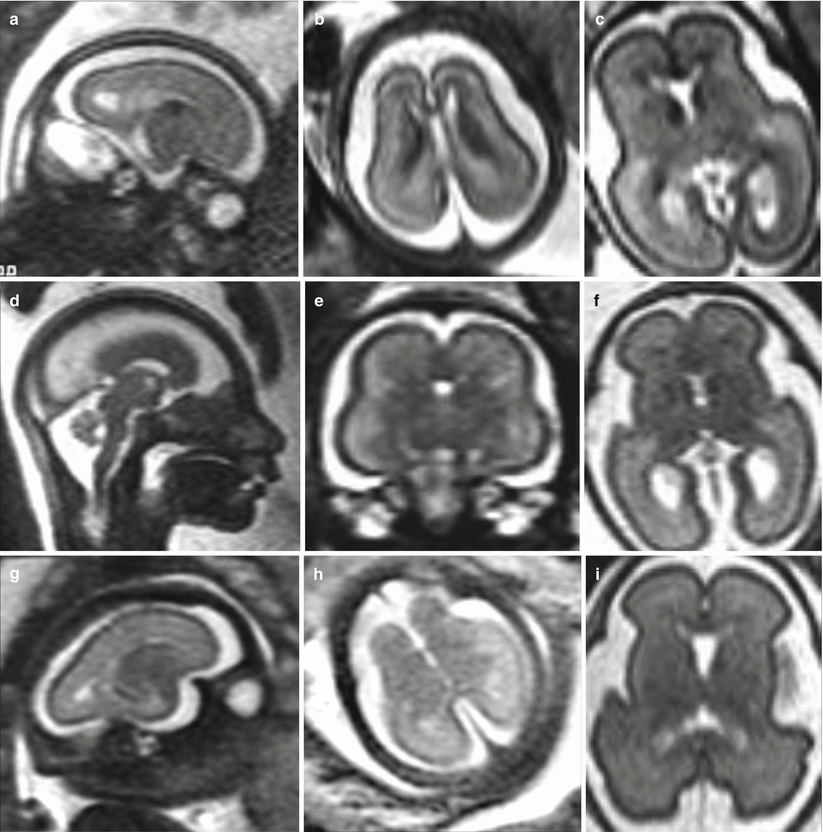
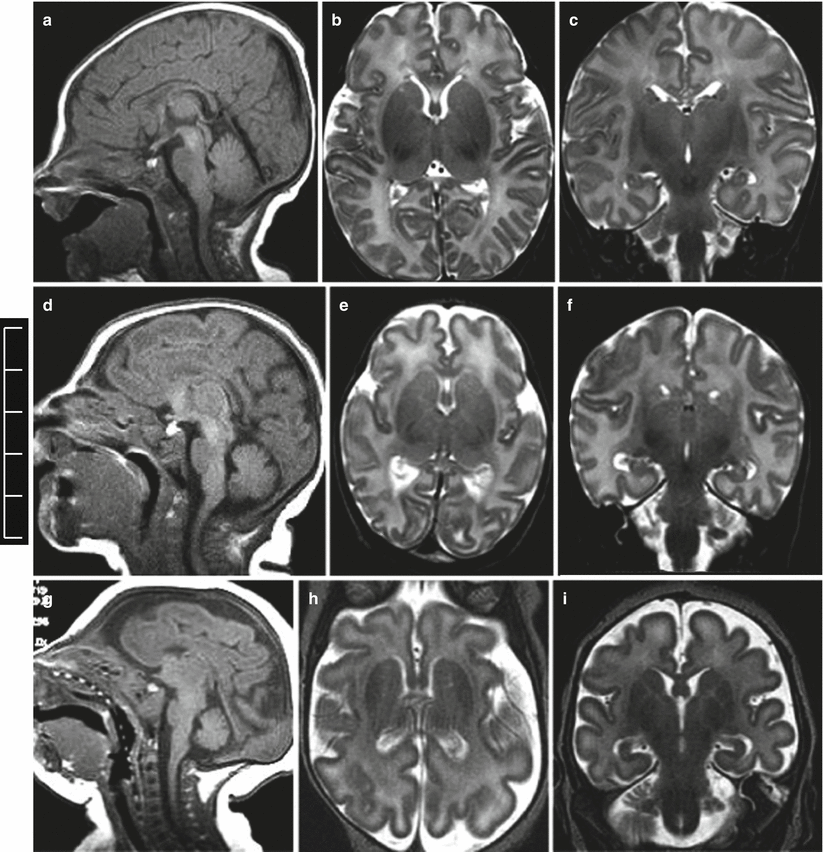
Fig. 4.1
Primary congenital microcephaly. MR fetal studies of microcephalic brain at 23 (a, b), 24 (d, e) and 27 (g, h) GW. The microcephalic fetal brain is compared with the normal brain for the correspondent GW (c, f, i)
Fig. 4.2
Primary congenital microcephaly. Neonatal MR studies, normal male brain with a head circumference of 35 mm (a–c), microcephalic male brain with a head circumference of 29 cm (d–f), microcephalic male brain with a head circumference of 26 cm (g–i)
4.2.2 Megalencephaly
Megalencephaly (MEG) can be grouped in two main conditions, MEG with or without polymicrogyria (PMG). MEG with an apparently normal cortex can be associated with some syndromes such as Sotos syndrome, Bannayan–Golabi–Behmel syndrome, or Proteus syndrome [1]. A fetal megalencephalic brain has been demonstrated on fetal MR also in a case of Alexander disease as a rare and severe metabolic disorder typically associated with macrocephaly [7].
MEG with PMG has been demonstrated in two syndromes, the macrocephaly, polymicrogyria, polydactyly, hydrocephalus syndrome (MPPH) [8] and the macrocephaly capillary syndrome (MCAP) [8, 9]. We reported a case of a megalencephalic brain in a fetus with in vivo and ex vivo fetal MR studies [10]. The megalencephalic brain showed diffuse polymicrogyria and a distinctive overgrowth of frontal regions (Fig. 4.3). This case did not seem to present all the diagnostic criteria of the MPPH/MCAP spectrum; however, megalencephaly was reported in 6 % of cases of the largest series of polymicrogyria cases available up to now [11], and cases of MEG–PMG association other than MPPH/MCAP could be reasonably hypothesized.
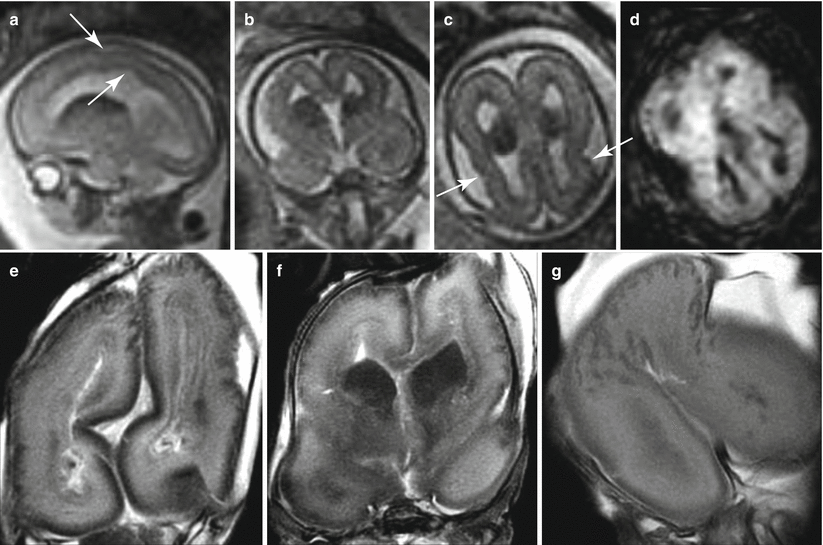
Fig. 4.3
Megalencephaly and polymicrogyria of unknown origin. Fetal MR (a–d) shows a dysmorphic megalencephalic brain with an overdevelopment of frontal lobes (larger than the parieto-occipital part of the brain). Some irregularities of cortical profile (arrows a, c) and ventricular margin (arrow a) can be seen. A large ganglionic eminence with a diffusion restriction (d) is detectable. Fetal MR autopsy (e–g) confirms the presence of large areas of microgyric appearance of the cortical plate
4.2.3 Cortical Dysgenesis with Abnormal Cell Proliferation (Without Neoplasia)
This group of MCD consists of (1) diffuse cortical dysgenesis and (2) focal and multifocal cortical dysgenesis.
The first group refers up to now to a single rare syndrome characterized by polyhydramnios, MEG, and symptomatic epilepsy (PMSE). The second group is larger and phenotypically heterogeneous; it includes hemimegalencephaly (HMEG) isolated and associated with neurocutaneous syndrome, focal cortical dysplasia (FCD) type IIa and IIb, and tuberous sclerosis.
HMEG is a rare and severe malformation characterized by an excessive growth of all or part of one hemisphere with dysplastic cortex (often polymicrogyric) and white matter abnormal signal. In some case the cerebellum may be involved. During pregnancy this malformation may be suspected when an asymmetry of the two hemispheres and lateral ventricles is detected by US. Very little information is available about fetal MR imaging in HMEG [12, 13]. The main findings are an enlargement of the affected hemisphere with ipsilateral ventricle dilatation and abnormal sulcation. A characteristic aspect of the fetal imaging is a ganglionic eminence/basal ganglia region hypertrophy characterized by hypointensity on T2-weighted images and low ADC value, suggestive for abnormally high cell density [12].
No cases of FCD type IIa or IIb are at present identified by means of fetal MR, and the fetal features of this malformation are still unclear.
Differently from FCD, fetal MR can diagnose in utero the brain involvement in cases of suspected tuberous sclerosis. Tuberous sclerosis complex (TSC) is an autosomal dominant phakomatosis caused by mutations in either the TSC1 (localized to chromosome 9q34) or TSC2 (localized to chromosome 16p13.3), tumor suppressor genes [14] It is characterized by widespread development of hamartomas in different organs in addition to brain abnormalities. During the prenatal period, the detection of cardiac rhabdomyomas on echocardiography is a strong predictor of TSC, and it is the indication to perform fetal brain MR imaging study even in the presence of normal fetal neurosonography. Fetal MR imaging is particularly useful for the detection of associated brain lesions specific of TSC that might support the diagnosis of the disease [15]. Brain lesions are responsible for severe clinical manifestations, such as neurobehavioral disabilities and intractable epilepsy, and their presence on fetal MRI has a great importance on the prenatal counseling. On histopathology the lesions are mainly composed of large bizarre cells called balloon cells scattered throughout the cortex and subcortical white matter. Among cerebral lesions, subependymal nodules are the most commonly reported findings prenatally (Figs. 4.4 and 4.5) [16]. Subependymal nodules have been identified as early as 21 weeks of gestation by fetal MR imaging; their number may increase during pregnancy [17]. They appear as nodules located at the edges of the lateral ventricles and deforming the ventricular wall hypointense on T2-weighted images and hyperintense on T1-weighted images (Fig. 4.5). Cortical tubers and white matter linear alterations (Figs. 4.5 and 4.6) may be more difficult to detect, mainly at early GW when radiological findings are very subtle [18]. Multiplicity of lesions increases the probability of TSC diagnosis [19]. It is nevertheless important to underline that normal prenatal MRI does not exclude definitively the diagnosis of TSC, and false-negative fetal cases are reported especially at early gestational age (Fig. 4.6). Fetal MR imaging follow-up studies may be therefore useful to reduce the risk of a missed diagnosis.
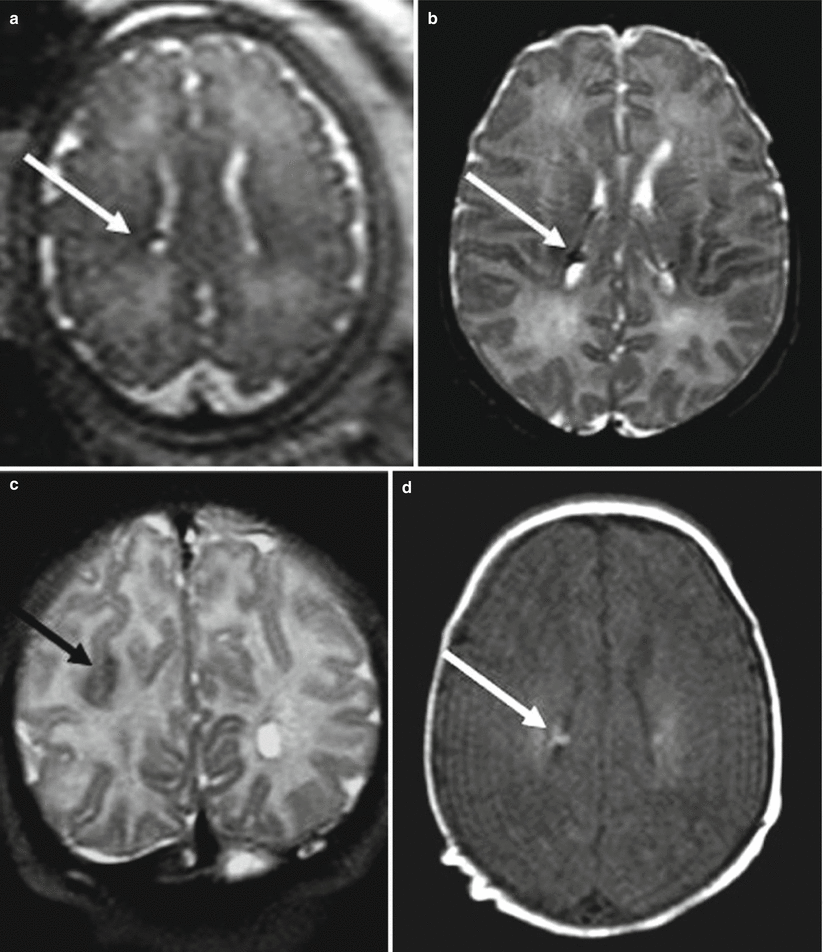
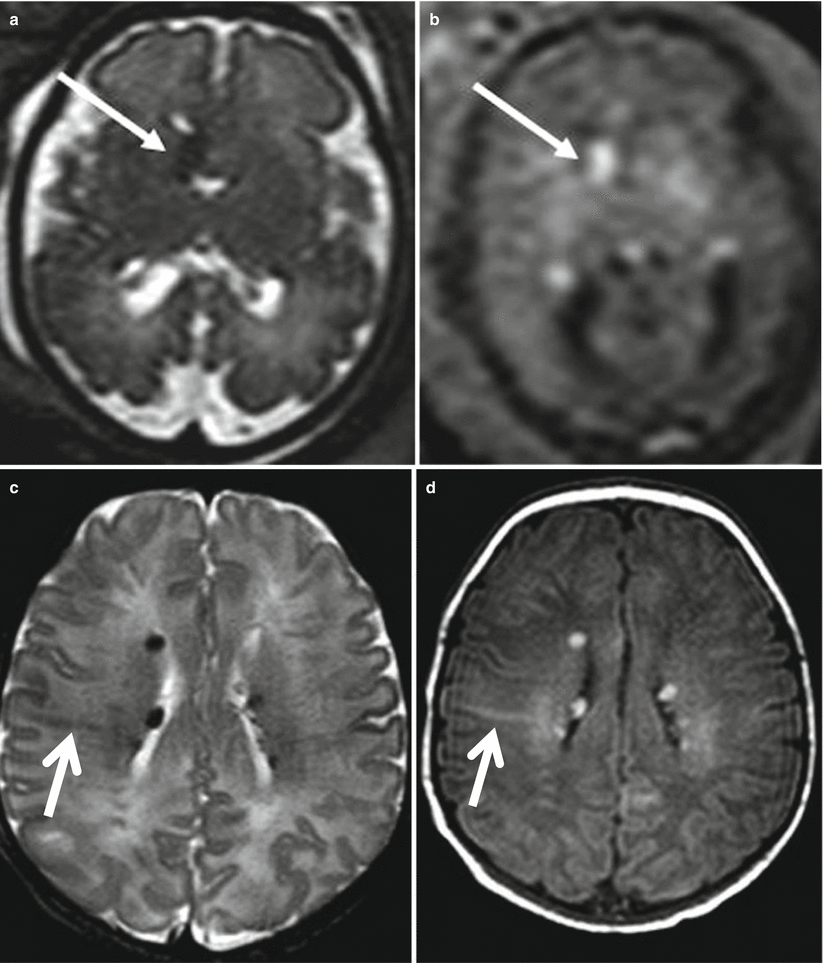
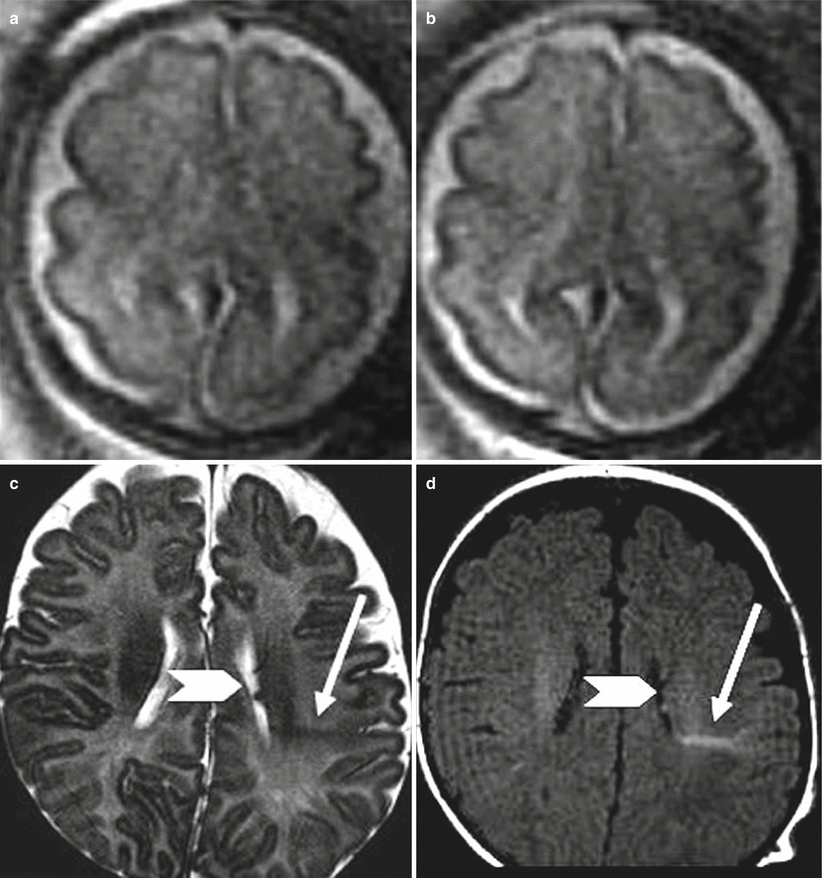
Fig. 4.4
Tuberous sclerosis. Fetal MR study at 34 GW (a) in a fetus with a cardiac rhabdomyoma shows a subependymal nodule (arrow). Postnatal MR study confirms the presence of a subependymal nodule hypointense on T2-weighted image (b) and hyperintense on T1-weighted image (d), a parietal tuber is evident as well on the postnatal study (c)
Fig. 4.5
Tuberous sclerosis. Fetal MR study at 31 GW (a, b) in a fetus with cardiac rhabdomyoma shows a subependymal nodule (arrow) near the foramen of Monro hypointense on T2-weighted image (a) and hyperintense on T1-weighted image (b), postnatal MR study (c, d) shows multiple subependymal nodules and white matter linear alteration (arrows)
Fig. 4.6
Tuberous sclerosis, fetal MR false-negative study. Fetal MR study at 27 GW (a, b) in a fetus with cardiac rhabdomyoma was referred as normal. Postnatal MR study (c, d) shows multiple subependymal nodules (arrowhead) and white matter linear alteration (arrows)
Congenital subependymal giant cell astrocytomas (SEGA) are very rare in fetal and neonatal period, and only a limited number of cases are reported in the literature. However, neonates with SEGA appear to have a particularly poor prognosis. SEGA are located near the foramen of Monro, they are greater than 10 mm in diameter and exhibit growth on serial imaging and may cause obstructive hydrocephalus [14, 20, 21].
4.3 Malformations due to Abnormal Cortical Migration
4.3.1 MCD with Neuroependymal Abnormalities: Periventricular Heterotopia
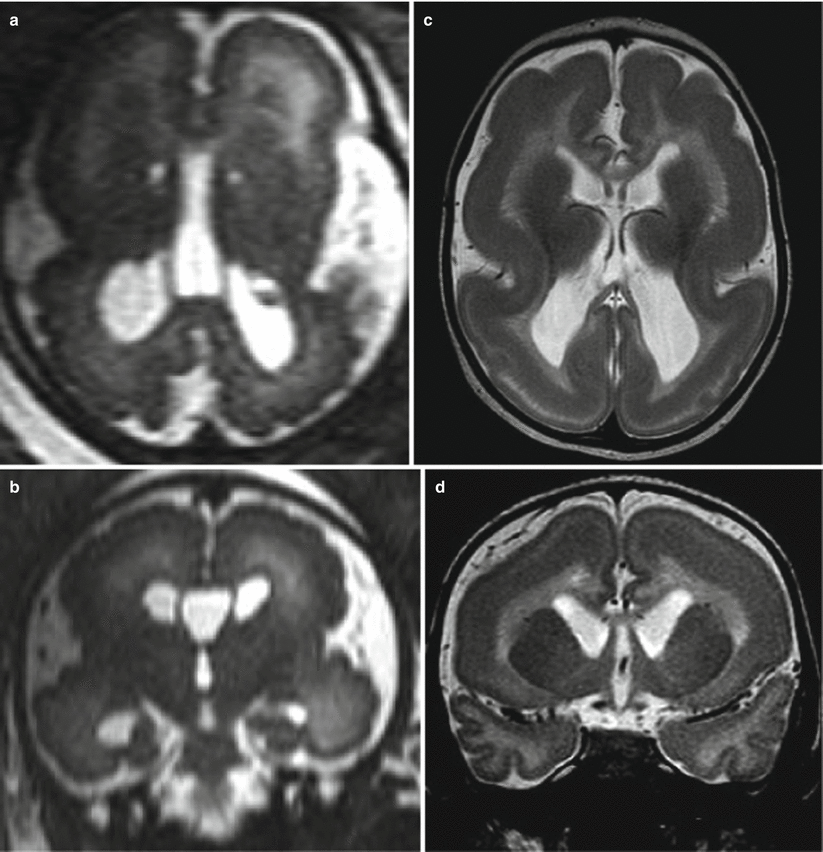
Periventricular nodular heterotopia (PNH) is a collection of nodules of gray matter consisting of clusters of neurons that fail to migrate, found along subependymal–periventricular region protruding into the ventricular lumen. In the classical form the PNH is diffuse, bilateral, and symmetric without other brain abnormalities, but the most common pattern of PNH is the bilateral posterior one (temporo-trigonal), frequently associated with developmental brain anomalies and polymicrogyric overlying cortex [22, 23]. In other cases PNH may have different localization with bilateral or unilateral symmetric or asymmetric distribution. PNH is usually not detected by prenatal US and represents an unexpected finding on fetal MRI (Fig. 4.7). During midgestation PNH features on fetal MRI may be very subtle presenting as a fine irregularity of ventricles walls; at late gestation PNH is more easily identified as low signal micronodules on T2-weighted images. Since PNH may be associated with other brain malformation and MCD, the overlying cortex has to be carefully evaluated (Fig. 4.8). On the other hand its worth to remind that we have experienced also false-positive cases of PNH.
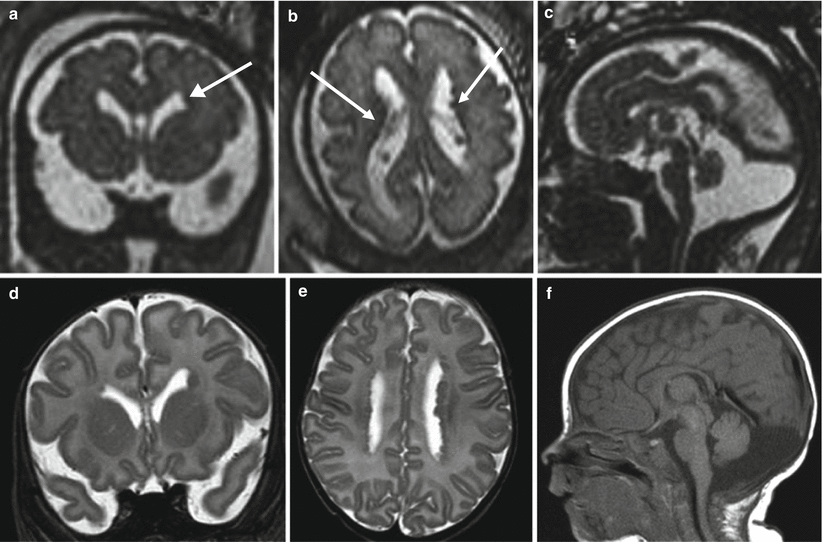
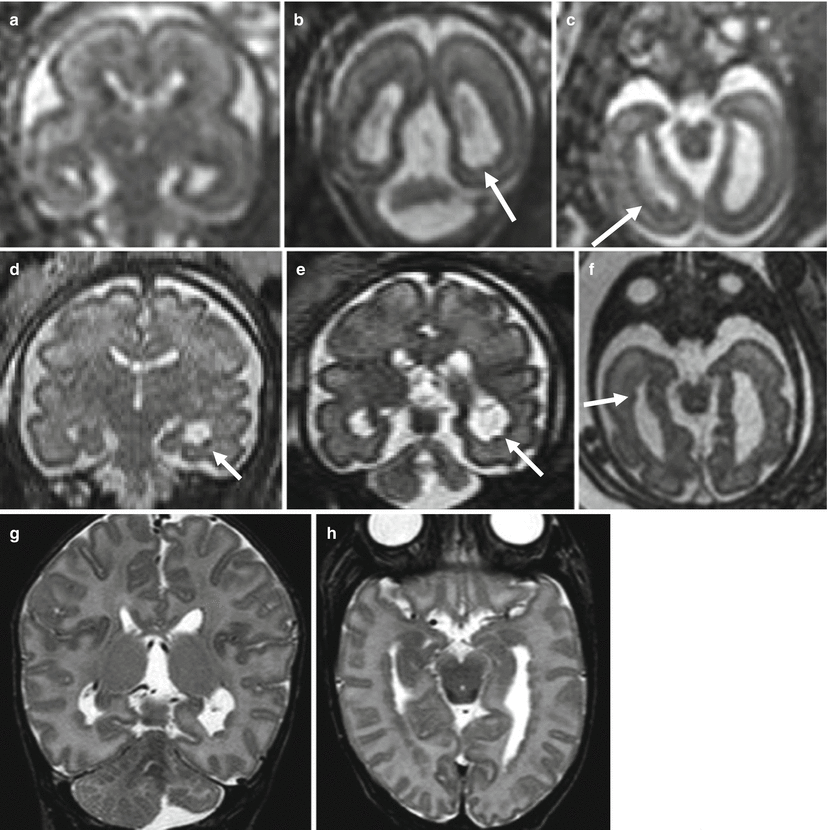
Fig. 4.7
PNH. Fetal MR at 30 GW (a–c) and postnatal MR (d–f) show bilateral periventricular nodularity (arrows a, b). This case was referred from US study for an enlargement of the cisterna magna that was confirmed both in pre- and postnatal studies
Fig. 4.8
PNH. Fetal MR at 21 (a–c) and 30 (d–f) GW 21. Small subependymal heterotopic nodules are evident on both prenatal studies (arrows) and confirmed in the postnatal MR (g, h). Left cerebellar hemisphere is reduced in size in the first study, and the hypoplastic aspect becomes progressively more evident in the second and third studies
PNH could be theoretically confused with hemosiderinic deposits, but multiple micronodules are likely to represent a malformative aspect more than a hemorrhagic sequelae, and in case of a single nodule, the heterotopic nodule is generally iso- or only slight hyperintense on T1-weighted images, while subependymal blood clots have usually a more marked bright T1 signal (Fig. 4.9). Another differential diagnosis is with subependymal nodules of tuberous sclerosis, but in these cases the strong association with cardiac rhabdomyomas detected on prenatal US allows a correct diagnosis to be made, and in addition nodules of tuberous sclerosis are mainly localized at the level of the foramen of Monro.
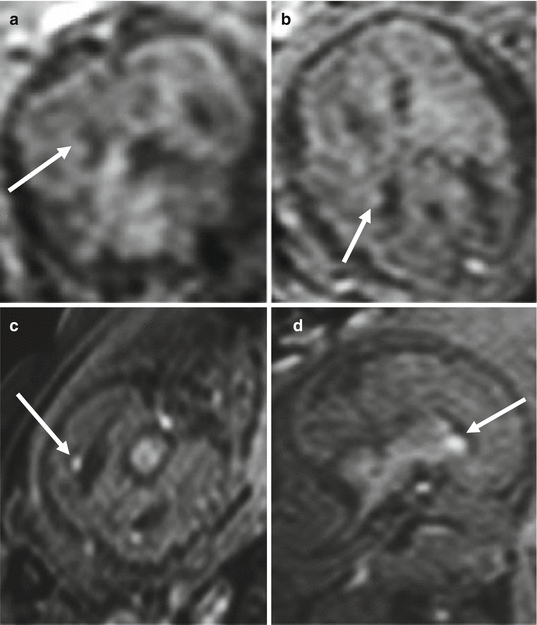
Fig. 4.9
PNH, differential diagnosis. T1-weighted image of the previous case at 21 GW (a, b) compared with subependymal clots at 25 GW (c, d). Subependymal clots (arrows c, d) are more hyperintense on T1-weighted images than heterotopic nodules (arrows a, b)
The detection and the differential diagnosis of PNH is usually easier on postnatal MR, and other findings associated with a possible previous hemorrhagic event or a neurocutaneous syndrome may be better visualized at this time.
4.3.2 MCD due to Generalized Abnormal Transmantle Migration Lissencephaly/Subcortical Band Heterotopia (LIS/SBH)
Lissencephaly (LIS) is the most severe form of failure of neuronal migration in which the cortical surface is smooth and brain volume is strongly reduced. The absence of cerebral gyri may be complete (agyria) or incomplete (pachigyria); in the latter case few, large and thick gyri are present. The thick cortex is composed by a thin outer cortical layer, a bright white matter layer called cell-sparse zone, and a thick inner band of neurons (deep cortex) [24]. Incomplete opercularization and a brain appearance of a figure of eight are typical. LIS has a strong genetic basis in which several genes, likely involved in regulation of microtubule activities, are implicated [25–27]. Classic LIS may be a part of Miller–Dieker syndrome (17p13.3 deletion) or Norman–Robert syndrome associated with facial dysmorphisms. Even rare these syndromes are recognized on prenatal images because of an almost total lack of sulci, mild ventriculomegaly, extreme microcephaly [28], and mainly facial anomalies such as micrognathia.
When not associated to other abnormalities, LIS may be difficult to identify on MR imaging at midgestation due to the physiological smooth aspect of the fetal brain; however, some diagnostic clues can be recognized also at this stage and first of all the absence of the expected fissures with shallow appearance of the sylvian fissure. This aspect may be seen as early as 21 GW. Other brain anomalies are frequently associated with LIS, such as dysmorphic aspect of the ventricles and various degrees of the corpus callosum, brainstem, and cerebellar hypoplasia may be present (Fig. 4.10).