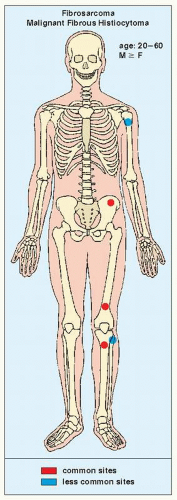
 FIGURE 22.1 Skeletal sites of predilection, peak age range, and male-to-female ratio in fibrosarcoma and MFH. |
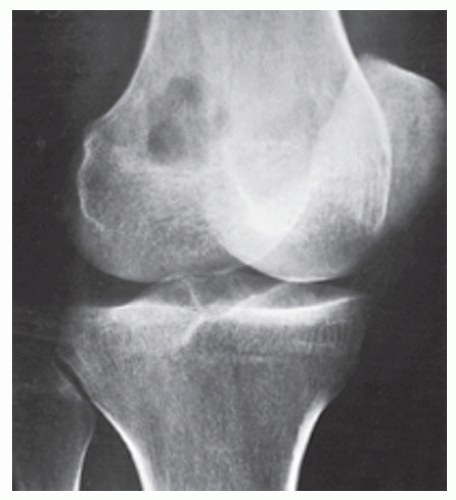 FIGURE 22.2 Fibrosarcoma. Oblique radiograph of the right knee of a 28-year-old woman shows a purely destructive osteolytic lesion in the intercondylar fossa of the distal femur. Note the absence of reactive sclerosis and periosteal response. |
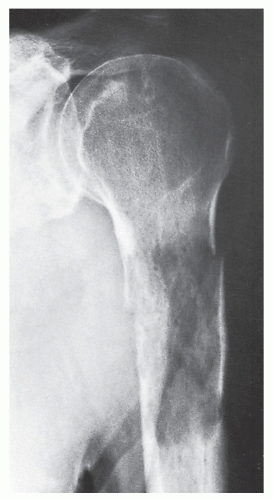 FIGURE 22.3 Fibrosarcoma. A 62-year-old man sustained a pathologic fracture through an osteolytic lesion in the proximal shaft of the left humerus. A metastatic lesion was suspected, but biopsy revealed a primary fibrosarcoma of the bone. |
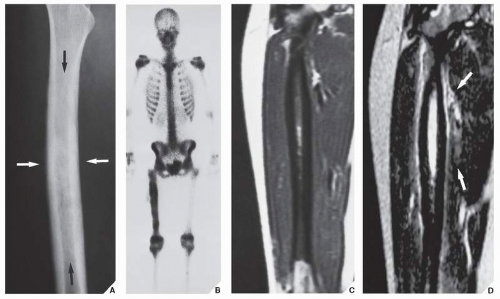 FIGURE 22.4 Malignant fibrous histiocytoma. (A) Oblique radiograph of the right femur of a 16-year-old girl shows fusiform thickening of the cortex and permeative type of medullary bone destruction (arrows). (B) Radionuclide bone scan (99mTc-MDP) shows increased uptake of the tracer in the right femur. (C) Coronal T1-weighted (SE; TR 500/TE 20 msec) MR image demonstrates the extent of the tumor that involves about 75% of the length of the femur. (D) Coronal T2-weighted (SE; TR 2000/TE 80 msec) MR image shows that the tumor exhibits high signal intensity. The soft-tissue extension medially is also accurately depicted (arrows). |
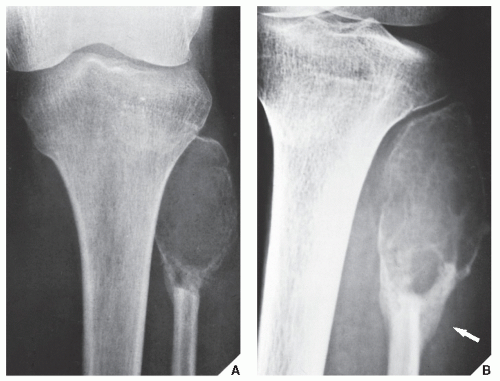 FIGURE 22.5 Malignant fibrous histiocytoma. Anteroposterior radiograph of the left knee (A) and oblique projection (B) demonstrate an expansive, lytic lesion in the proximal end of the fibula in a 13-year-old girl. The cortex has been partially destroyed, and there is a buttress of periosteal new bone formation (arrow) secondary to pathologic fracture. The differential diagnosis of this malignancy at this site should include giant cell tumor and aneurysmal bone cyst. |
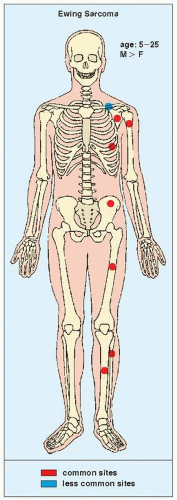 FIGURE 22.6 Skeletal sites of predilection, peak age range, and male-to-female ratio in Ewing sarcoma. |
this lesion usually occurs in an older age group. The important radiologic difference is usually the absence of a soft-tissue mass in lymphoma, whereas in Ewing sarcoma a soft-tissue mass is almost invariably present, often being disproportionally large compared with the amount of bone destruction (see Figs. 22.8 and 22.9). The distinction between Ewing sarcoma and PNET cannot be made on the basis of radiography. Differentiation between these two tumors must rely entirely on immunohistochemistry, electron microscopy, and molecular genetic studies.
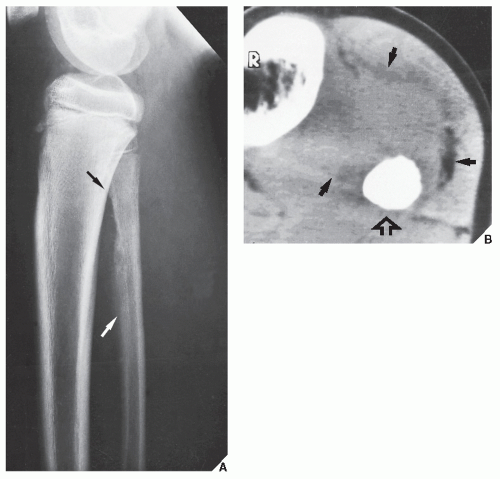 FIGURE 22.7 Ewing sarcoma. (A) Lateral radiograph in a 12-year-old boy shows the typical appearance of this tumor in the fibula. The poorly defined lesion exhibits permeative bone destruction associated with an aggressive periosteal reaction (arrows). (B) CT section through the lesion demonstrates a large soft-tissue mass (arrows), which is not clear on the conventional study. Note the complete obliteration of the marrow cavity by tumor (open arrow). |
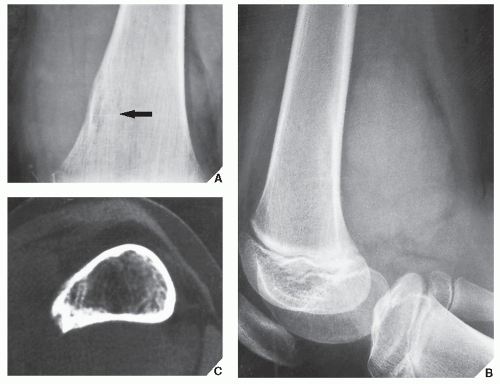 FIGURE 22.8 Ewing sarcoma. (A) Bone destruction (arrow) is almost imperceptible on this magnification study in a 10-year-old girl with Ewing sarcoma of the distal femoral diaphysis. (B) Lateral radiograph of distal femur, however, shows a large soft-tissue mass. (C) CT using a bone “window” demonstrates destruction of the medullary portion of the bone, endosteal scalloping, and invasion of the cortex. |
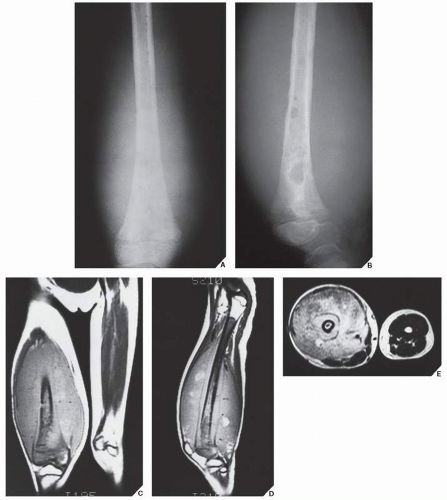 FIGURE 22.9 MRI of Ewing sarcoma. Anteroposterior (A) and lateral (B) radiographs of the right distal femur of a 7-year-old girl show permeative and moth-eaten types of bone destruction in the metaphysis and diaphysis associated with a large soft-tissue mass. Coronal (C) and sagittal (D) T1-weighted (SE; TR 750/TE 20 msec) MR images demonstrate the intraosseous and extraosseous extent of the tumor. (E) Axial T2-weighted (SE; TR 2000/TE 80 msec) MR image shows heterogeneous but mostly high signal intensity of the soft-tissue mass. |
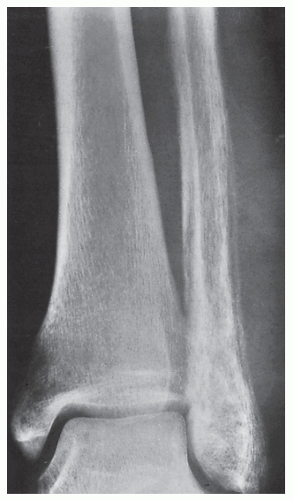 FIGURE 22.10 Ewing sarcoma. A 24-year-old man presented with pain and swelling of the left ankle for 8 weeks; he also had a fever. Anteroposterior radiograph of the ankle demonstrates a destructive lesion of the distal fibula exhibiting a permeative type of bone destruction and a lamellated periosteal reaction; a soft-tissue mass is also evident. The appearance is that of infection (osteomyelitis), but biopsy confirmed malignancy. |
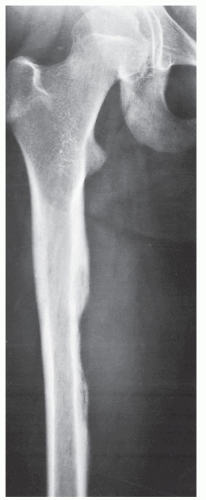 FIGURE 22.11 Ewing sarcoma. Anteroposterior radiograph of the right femur of a 12-year-old girl shows “saucerization” of the medial cortex of the diaphysis, often seen in Ewing sarcoma; there is also an associated soft-tissue mass. |
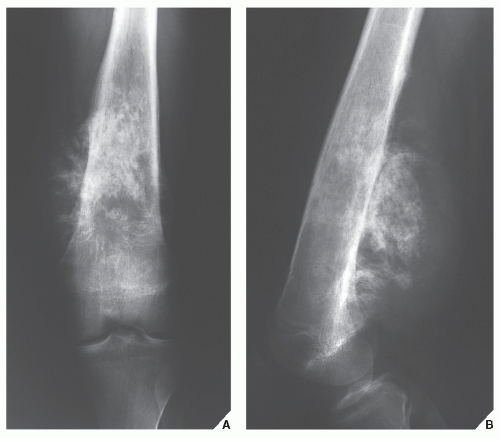 FIGURE 22.12 Ewing sarcoma. Anteroposterior (A) and lateral (B) radiographs of the left femur of a 17-year-old boy show a tumor displaying a significant degree of sclerosis that was originally interpreted as osteosarcoma. |
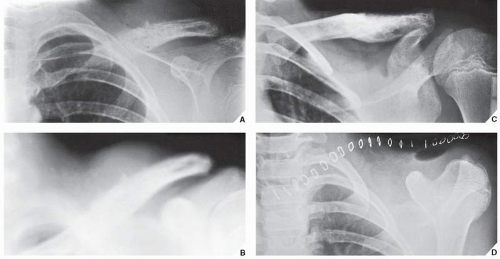 FIGURE 22.13 Treatment of Ewing sarcoma. (A) Radiograph of the shoulder of an 11-year-old boy shows the typical appearance of Ewing sarcoma involving the distal half of the left clavicle. The poorly defined destructive lesion is associated with an aggressive periosteal reaction and a large soft-tissue mass. (B) Tomographic cut gives a better picture of the periosteal response and soft-tissue mass. (C) After a 4-month course of chemotherapy, the lesion has become sclerotic, the periosteal reaction has disappeared, and the soft-tissue mass has shrunk substantially. (D) The clavicle was then removed en bloc. |
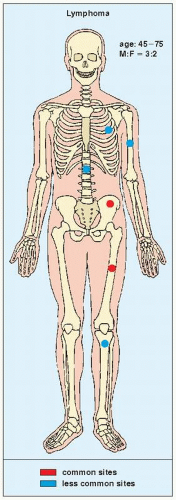 FIGURE 22.14 Skeletal sites of predilection, peak age range, and male-to-female ratio in primary bone lymphoma. |
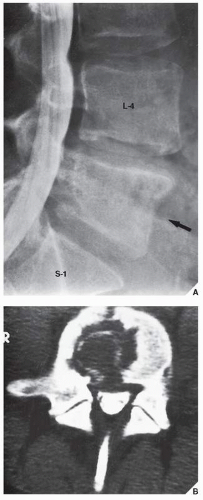 FIGURE 22.15 Lymphoma. An 18-year-old woman presented with low-back pain for several months, which was attributed to herniation of an intervertebral disk. (A) Myelogram shows that the disk is normal, but the body of L5 (arrow) exhibits a mottled appearance and its posterior border is indistinct. (B) CT section demonstrates a large, osteolytic lesion extending from the anterior to the posterior margins of the vertebral body. |
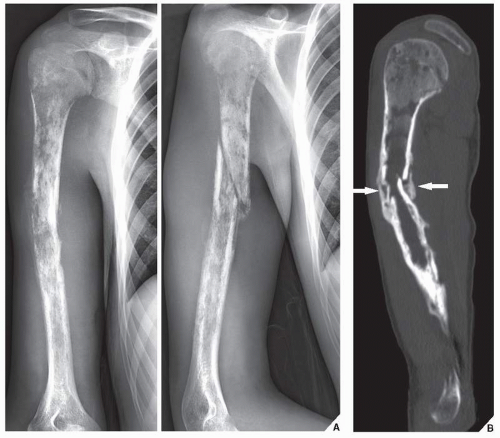 FIGURE 22.16 Lymphoma. (A) Anteroposterior and oblique radiographs of the right humerus of a 20-year-old man show a long lesion exhibiting permeative and moth-eaten type of bone destruction. Periosteal reaction is secondary to the pathologic fracture. (B) Sagittal reformatted CT image demonstrates endosteal scalloping and early callus formation at the site of a pathologic fracture (arrows). |
TABLE 22.1 Revised European-American Lymphoma Classification | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
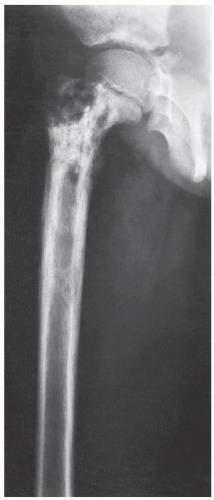 FIGURE 22.17 Lymphoma. Conventional radiograph of the right femur of a 7-year-old girl with groin pain and a fever reveals a destructive lesion of the diaphysis extending to the growth plate; there is also a lamellated type of periosteal reaction. Because of the age of the patient, the primary differential diagnosis included Ewing sarcoma, osteomyelitis, and Langerhans cell histiocytosis, all three of which may have a similar radiographic presentation in a long bone. The main factor differentiating these lesions is the duration of the patient’s symptoms. In this case, however, biopsy revealed a histiocytic lymphoma. |
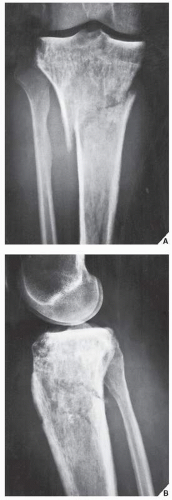 FIGURE 22.18 Lymphoma. Anteroposterior (A) and lateral (B) radiographs of the right knee of a 47-year-old woman who had knee pain and initially was misdiagnosed with Paget disease, show a destructive lesion of the proximal tibia extending into the articular end of the bone. The mixed sclerotic and osteolytic character of this lesion may resemble the coarse trabecular pattern of Paget disease; however, there is a lack of cortical thickening. There is a pathologic fracture, but only a minimal periosteal response is evident. |
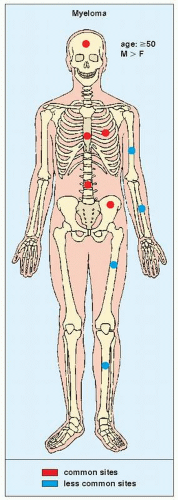 FIGURE 22.19 Skeletal sites of predilection, peak age range, and male-to-female ratio in myeloma. |
fractures of the vertebral bodies may also be evident. More commonly, it exhibits multiple lytic lesions scattered throughout the skeleton. In the skull, characteristic “punched-out” areas of bone destruction, usually of uniform size, are noted (Fig. 22.21), whereas the ribs may contain lace-like areas of bone destruction and small osteolytic lesions, sometimes accompanied by adjacent soft-tissue masses. Areas of medullary bone destruction are noted in the flat and long bones, and if these appear about the cortex, they are accompanied by scalloping of the inner cortical margin (Fig. 22.22). Ordinarily, there is no evidence of sclerosis and no periosteal reaction. Fewer than 1% of myelomas may be of a sclerosing type called sclerosing myelomatosis.
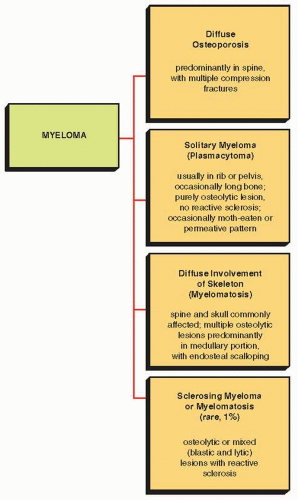 FIGURE 22.20 Variants in the radiographic presentation of myeloma. |
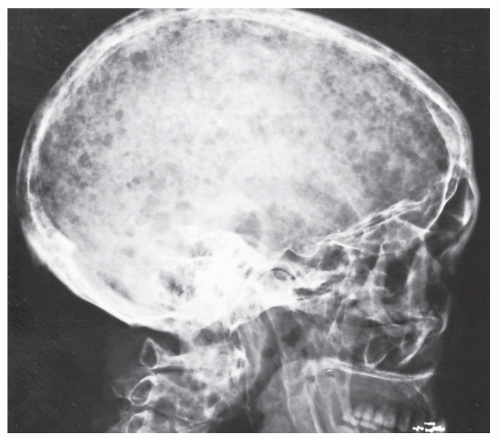 FIGURE 22.21 Multiple myeloma. Involvement of the skull is prominent in this 60-year-old woman. Note the characteristic “punched-out,” lytic lesions, most of which are uniform in size and lack sclerotic borders. Occasionally, this pattern may be seen in metastatic disease. |
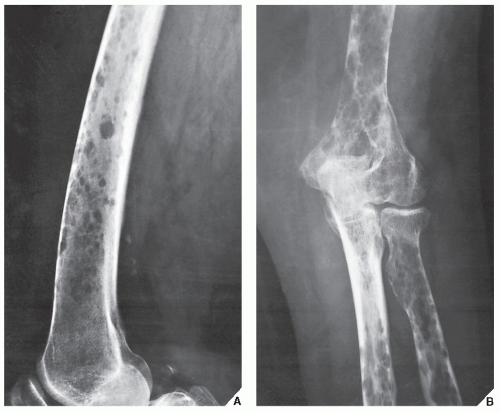 FIGURE 22.22 Multiple myeloma. Lateral radiograph of the distal femur (A) and anteroposterior radiograph of the elbow (B) in a 65-year-old woman show endosteal scalloping of the cortex typical of diffuse myelomatosis. |
myeloma, however, both the pedicle and vertebral body may be destroyed. Radionuclide bone scan can more reliably distinguish these two malignancies at this stage. It is invariably positive in cases of metastatic carcinoma, whereas in most cases of multiple myeloma there is no increased uptake of radiopharmaceutical tracer. This phenomenon appears to reflect the purely lytic nature of most myelomatous lesions and the absence of significant reactive new bone formation in response to the tumor.
 FIGURE 22.23 Multiple myeloma versus metastatic carcinoma. Anteroposterior (A) and lateral (B) radiographs of the spine in a 70-year-old man with multiple myeloma involving both the spine and appendicular skeleton show a compression fracture of the body of T8; several other vertebrae show only osteoporosis. The pedicles are preserved in contrast to metastatic disease of the spine, which usually also affects the pedicles, as seen on this anteroposterior radiograph of the cervical spine (C) in a 65-year-old man with colon carcinoma and multiple lytic metastases. Note the involvement of the right pedicle of C7 (open arrows). |
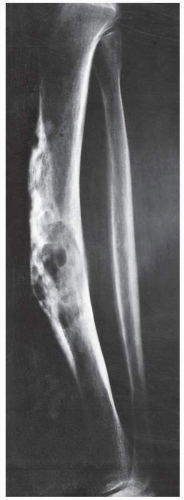 FIGURE 22.24 Adamantinoma. Lateral radiograph of a 64-year-old woman shows a lesion in the midshaft of the left tibia. The destructive lesion is multifocal and slightly expansive, with mixed osteolytic and sclerotic areas creating a “soap-bubble” appearance resembling that of osteofibrous dysplasia (see Fig. 19.40). |
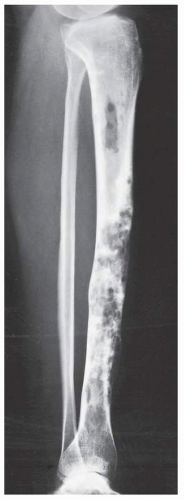 FIGURE 22.25 Adamantinoma. Lateral radiograph of the right leg of a 28-year-old woman shows multiple, confluent lytic lesions involving almost the entire tibia; only the articular ends are spared. The anterior cortex exhibits a predominantly “sawtooth” type of destruction. |
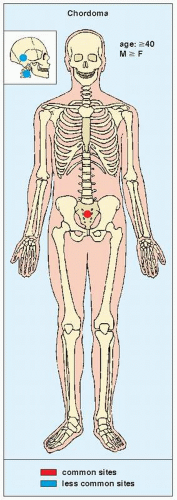 FIGURE 22.26 Skeletal sites of predilection, peak age range, and male-to-female ratio in chordoma. |
Related posts:
 Radiologic Evaluation of Skeletal Anomalies
Radiologic Evaluation of Skeletal Anomalies
 Inflammatory Arthritides
Inflammatory Arthritides
 Benign Tumors and Tumor-like Lesions II: Lesions of Cartilaginous Origin
Benign Tumors and Tumor-like Lesions II: Lesions of Cartilaginous Origin
 Upper Limb III: Distal Forearm, Wrist, and Hand
Upper Limb III: Distal Forearm, Wrist, and Hand
 Benign Tumors and Tumor-Like Lesions IV: Miscellaneous Lesions
Benign Tumors and Tumor-Like Lesions IV: Miscellaneous Lesions
 Upper Limb III: Distal Forearm, Wrist, and Hand
Upper Limb III: Distal Forearm, Wrist, and Hand
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


