Fig. 1
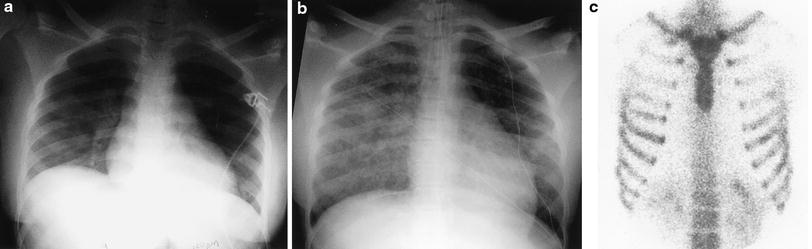
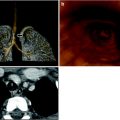
Juvenile idiopathic arthritis (JIA). A frontal CXR demonstrates a large pericardial effusion and a small left pleural effusion in an 8-year-old boy with JIA
2.2 Juvenile Systemic Sclerosis
Scleroderma is characterized by fibrotic infiltration of connective tissues. Involvement of the skin and the gastrointestinal tract, particularly the esophagus, is most common. The term systemic sclerosis is used to describe disseminated disease. Systemic sclerosis presents most commonly in adult women, but 10 % of cases present in children as juvenile systemic sclerosis (JSS). Both PFT abnormalities and lung parenchymal abnormalities on imaging are common in JSS. In a study of 11 JSS patients aged 5–19 years, 8 had ILD on CT. The most common abnormalities were peripheral ground-glass opacities in eight, subpleural nodules in seven, peripheral reticular opacities in six, and honeycombing in five (Seely et al. 1998) (Fig. 2). In a more recent and much larger study of 153 JSS patients, the prevalence of ILD on CT was 5 % at diagnosis and 23 % at some point in the overall course. Pulmonary hypertension occurred in 7 % (Martini et al. 2006). The severity of CT abnormalities correlates inversely with FEV1 (forced expiratory volume in one second), FVC (forced vital capacity), and DLCO (lung diffusion capacity for carbon monoxide) values (Panigada et al. 2009). Pulmonary hypertension can develop without lung parenchymal disease, possibly due to direct pulmonary vascular involvement (Cheema and Quismorio 2001).
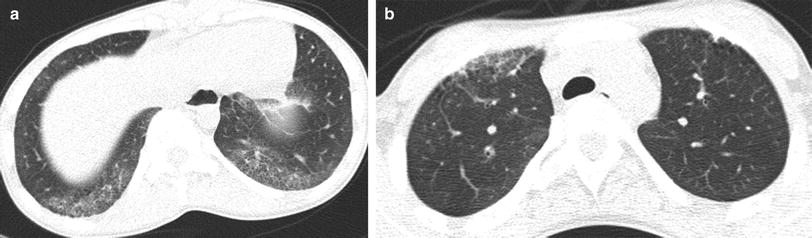
Fig. 2
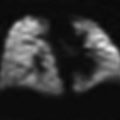
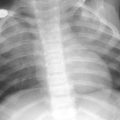
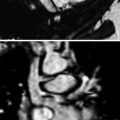
Juvenile systemic sclerosis (JSS). Axial chest CT images (a, b) from a 17-year-old girl with JSS show ground-glass opacities, fine reticular opacities, and tiny cysts at the peripheral posterior lower lobes and peripheral anterior upper lobes, as well as esophageal dilation
Although frequently described as honeycombing, the peripheral reticular opacities and cysts that are commonly seen in JSS often do not meet strict diagnostic criteria for honeycombing, and there is poor interobserver agreement for the identification of honeycombing, even among expert thoracic radiologists. In our experience, unlike the case in idiopathic pulmonary fibrosis, these findings may remain stable for long periods of time, which is consistent with reports that there is no correlation between the duration of illness and the severity of the lung disease (Panigada et al. 2009). However, since pulmonary fibrosis is a leading cause of mortality and progression of fibrosis may be clinically silent, measuring serum levels of KL-6 (Vesely et al. 2004) or obtaining serial CT exams (Seely et al. 1998; Panigada et al. 2009) has been suggested to monitor ILD in JSS. Preliminary studies have shown that lung ultrasound findings correlate with CT findings and PFT results in adults with ILD related to systemic sclerosis (Delle Sedie et al. 2012). If validated in children, ultrasound could provide a means to monitor ILD in JSS patients without ionizing radiation exposure. Aspiration and infection must also be considered as potential causes of lung disease in JSS patients given the high rate of esophageal dysmotility and gastroesophageal reflux and the use of immunosuppressive therapy (Christmann et al. 2010).
Scleroderma sin scleroderma, a rare form of scleroderma, presents without a skin rash and may be difficult to diagnose. Usually an adult disease, it has been reported in children as young as 6 years of age. Lung involvement is reported in approximately two-thirds of these patients (Toya and Tzelepis 2009).
2.3 Juvenile Dermatomyositis
Abnormal PFTs are reported in more than more than half of patients with juvenile dermatomyositis (JDM) (Takizawa et al. 1987). In a report of five cases, interstitial pneumonia was noted in three and COP in two. All had abnormalities on physical examination or imaging studies at the time of presentation with JDM, although in two cases these abnormalities were initially mild (Kobayashi et al. 2003).
A more recent report of 21 pediatric patients found that respiratory involvement was common, occurring in 76 %, but respiratory symptoms were not the presenting symptoms in any. Respiratory muscle involvement was the most common abnormality followed by interstitial lung disease. Chest CT was abnormal in 12 of the 15 patients in whom CT was performed, with linear opacities, nodules, ground glass opacities, expiratory air trapping, and bronchial wall thickening being the most common findings (Pouessel et al. 2013). Rarely, JDM may be complicated by an acute, rapidly progressive, steroid-refractory, fatal interstitial lung disease (Lin et al. 2002).
2.4 Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a multisystem disease characterized by persistent B-cell activation and overproduction of autoantibodies and immune complexes. Both the activity of the autoantibodies and the deposition of the immune complexes are associated with organ dysfunction (Lehman 1995). SLE is more common in non-Caucasian than in Caucasian populations, and susceptibility to SLE is thought to be related to multiple genes. The number of SLE susceptibility alleles relates to the likelihood of onset in childhood in African-Americans, but not in populations of Hispanic or European origin (Webb et al. 2011). One-fifth to one-sixth of SLE patients present before the age of 16 years (Arkachaisri and Lehman 1999). A comparison of adult- and pediatric-onset SLE found that children tend to present with more acute illness and have higher mortality rates (Mina and Brunner 2010).
In 37 % of cases of childhood-onset SLE, the pulmonary function is impaired, with a reduction of DLCO most commonly noted. Pulmonary abnormalities on chest CT are less common, with a prevalence of 8 %, and pulmonary function does not correlate with CT findings (Lilleby et al. 2006). Manifestations of SLE on chest imaging include pleural effusion, pericardial effusion, pneumonitis, obliterative bronchiolitis, vasculitis, pulmonary hemorrhage and pulmonary embolism (Babyn and Doria 2005). Opportunistic infections are a leading cause of death in children with SLE, so infection should be excluded before attributing respiratory signs or symptoms to primary lupus involvement (Wang et al. 2003).
Acute lupus pneumonitis is a rare, life-threatening condition related to diffuse alveolar damage. Acute lupus pneumonitis can be difficult to diagnose due to its nonspecific presentation of shortness of breath and extensive pulmonary opacification that simulates infectious pneumonia or pulmonary edema (Vece and Fan 2010) (Fig. 3).
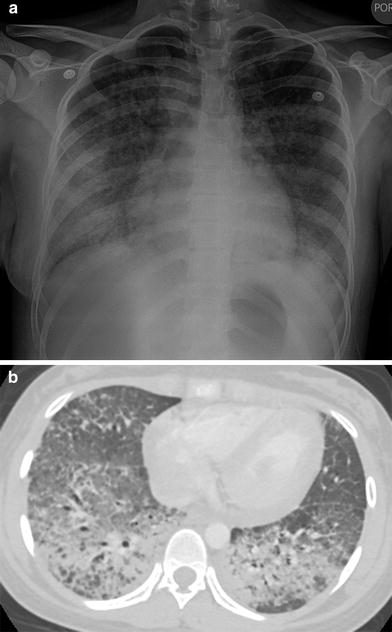
Fig. 3
Acute lupus pneumonitis. A frontal CXR (a) and an axial chest CT image (b) depict extensive bilateral pulmonary air space opacification in this 13-year-old girl with SLE and acute respiratory symptoms
Alveolar hemorrhage from SLE is more common in children than in adults, and the mortality is higher (Araujo et al. 2012). Patients present with a decrease in hematocrit, but hemoptysis may be absent. This condition can potentially be treated successfully with steroids and cytotoxic agents, so correct identification is important (Schwab et al. 1993). Air space opacities can be seen on CXR and CT. The imaging is not specific, as lupus pneumonitis or infection can produce the same findings. The finding of T2 shortening on magnetic resonance imaging (MRI) has been reported as a means of specifically identifying hemorrhage (Hsu et al. 1992).
“Shrinking lung syndrome” is a term used to describe a progressive decrease in lung volumes seen in some patients with SLE. This is usually identified on CXR as a progressive elevation of the diaphragm despite attempted full inspiration. The etiology is unknown, but may relate to a combination of pleural restriction due to recurrent pleural inflammation, pulmonary restriction due to pulmonary fibrosis, and weakness of the diaphragm and chest wall musculature. African-American patients are most commonly affected (Ferguson and Weinberger 2006).
3 Systemic Granulomatous Disorders
3.1 Sarcoidosis
Imaging findings of thoracic sarcoidosis in children are similar to those in adults. The most common thoracic manifestation of sarcoidosis in children is bilateral hilar lymphadenopathy. Pulmonary parenchymal disease occurs in approximately two-thirds of cases, usually in association with lymphadenopathy. Isolated pulmonary parenchymal involvement is found in only 11 % of cases (Keesling et al. 1998). Typical CT findings of pulmonary sarcoidosis in children include nodular beading and thickening of the bronchovascular bundles, interlobular septae, and fissures (Milman et al. 1998) (Fig. 4). Ground-glass opacities are also common, but parenchymal distortion and consolidation are less frequent and cysts or pleural effusions are typically not seen. Decreases in CT finding scores over time are associated with improvement of pulmonary function measured by FEV1 and FVC, so that the need for follow-up CT scans may be reduced when PFT results improve over time (Sileo et al. 2013a).
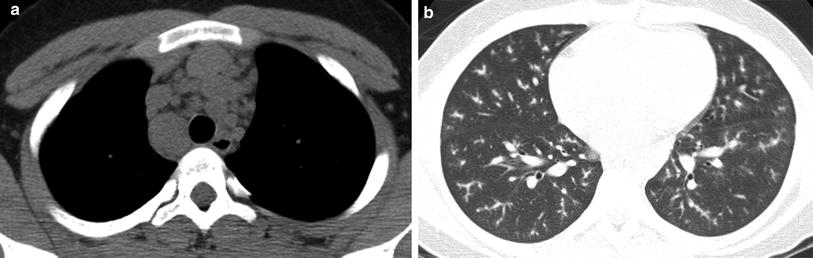
Fig. 4
Sarcoidosis. An axial chest CT image reconstructed in soft tissue windows (a) from a 13-year-old boy with sarcoidosis demonstrates mediastinal lymphadenopathy (a). An axial chest image reconstructed in lung windows (b) shows nodular beading of the pulmonary bronchovascular bundles
3.2 Crohn Disease
Although extraintestinal manifestations of Crohn disease are well known, respiratory tract involvement is uncommon but increasingly recognized, including in children (Al-Binali et al. 2003). Involvement can range from the large central airways (bronchiectasis, chronic bronchitis), to the small airways (bronchiolitis obliterans) and lung parenchyma (eosinophilic pneumonia, organizing pneumonia, granulomatous pneumonitis, interstitial pneumonitis) (Fig. 5). Drug-induced lung disease should be considered, particularly in the setting of eosinophilic pneumonia in patients on sulfasalazine or mesalamine therapy. Infection, including tuberculosis, should also be considered given the altered immune status of many of these patients. Treatment of pulmonary involvement is usually based on discontinuation of possible inciting drugs and initiation of steroids or other immunosuppressants (Basseri et al. 2010).
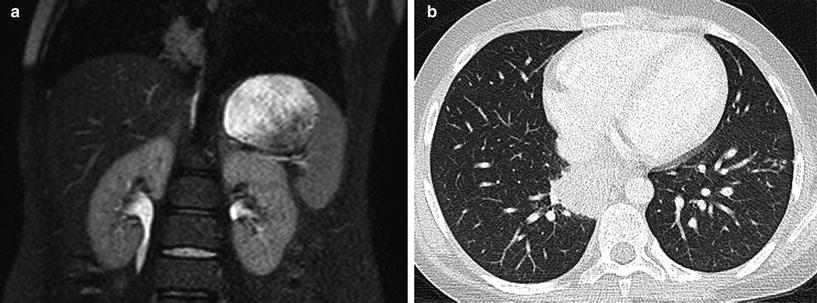
Fig. 5
Pulmonary Crohn disease. A coronal T2-weighted MR image (a) obtained during an MR enterography study on an 11-year-old boy with Crohn disease reveals a mass-like intermediate-to-high signal intensity lesion of the medial basilar right lower lobe. An axial chest CT image (b) shows a mass-like opacity of the medial basilar right lower lobe that was found to represent granulomatous inflammation on subsequent biopsy
4 Immunodeficiencies
Defences against infection include physical barriers, B-cells, T-cells, natural killer cells, phagocytes, and complement proteins. Defects in one or more of these components of the immune system result in an increased risk of infection. The lungs are exposed to both inhaled and circulating infectious agents and are frequently the site of infection in immunocompromised children.
Acquired immunodeficiency states are common in children, and can relate to chemotherapy for cancer, solid organ transplantation, hematopoietic stem cell transplantation (HSCT), immunosuppressive therapy for autoimmune disorders, or HIV infection. Although individually rare, the primary immunodeficiency diseases (PIDs) have a collective prevalence of 1/2,000 children in the United States (Boyle and Buckley 2007). Accumulating knowledge of the genetic and molecular basis of the immune system is providing a far more detailed understanding of PID, and specific genetic defects leading to distinct types of PID are increasingly being identified. With this increased understanding has come increased complexity in classifying PID. At least 200 inborn errors of immunity have been genetically defined, and the classification of PID is updated on a biennial basis by an expert committee of the International Union of Immunological Societies (IUIS). Based on disease mechanism, the IUIS classification groups PIDs into eight categories: predominantly antibody deficiencies, combined T-cell and B-cell immunodeficiencies, well-defined syndromes with immunodeficiency, congenital defects of phagocyte number and/or function, diseases of immune dysregulation, defects in innate immunity, auto-inflammatory disorders, and complement deficiencies. There is considerable heterogeneity, as well as overlap, in the phenotypic expression of these disorders (Al-Herz et al. 2011).
The radiologist has several roles when assessing the chest of children with an immunodeficiency. These include suggesting the possibility of an immunodeficiency, noting features characteristic of a specific immunodeficiency, evaluating infections, and detecting malignancies that can occur as a complication of certain immunodeficiencies.
By noting repetitive, severe, or refractory infections or recognizing certain patterns of disease, the radiologist may be the first to suggest the possibility of an immunodeficiency. In a recent study, 96 % of children with PIDs were referred to pediatric immunodeficiency centers by hospital clinicians rather than general pediatricians, illustrating the difficulty in making an early diagnosis of PID (Subbarayan et al. 2011). Early recognition of PID is important for several reasons. Bronchiectasis resulting from repeated or inadequately treated respiratory tract infection is irreversible. Outcomes are improved and treatment costs are reduced with institution of appropriate therapy, which can include immunoglobulin replacement therapy, prophylactic antimicrobials, and hematopoietic stem cell reconstitution. Avoidance of live vaccines and nonirradiated cellular blood products may be needed (Modell et al. 2009). Due to the heritable nature of many of these disorders, genetic testing and counseling may be warranted.
The primary role of thoracic imaging in children with immunodeficiencies is the detection and therapy response assessment of pulmonary infections. CXRs remain the most frequently obtained imaging study. However, CT scanning has long been recognized as both more sensitive and specific than CXR for the evaluation of diffuse infiltrative lung disease (Mathieson et al. 1989). CT is useful in children with antibody deficiency disorders to demonstrate the extent and severity of lung disease (Manson et al. 1997). Aerogenous spread of mycobacterial infection will frequently show a “tree-in-bud” appearance of infectious material filling dilated distal bronchioles (Fig. 6), although this appearance is far less specific than originally reported. Invasive aspergillosis may manifest as pulmonary nodules with a “halo” of ground-glass opacity (Seely et al. 1997) (Fig. 7). Pneumocystis jirovecii pneumonia (PCP) has a broad spectrum of findings that can include ground-glass opacities, reticulonodular opacities, septal thickening, and cysts (George et al. 2009) (Fig. 8). When identification of a specific etiology is required, CT can be used to guide bronchoscopy or needle biopsy to increase the yield of these procedures (Spencer et al. 1996).
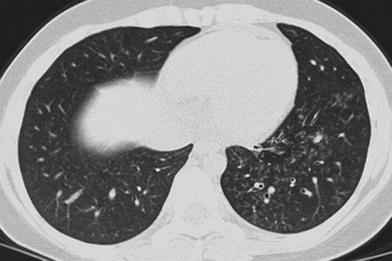
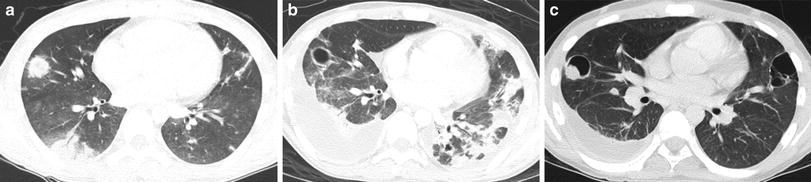
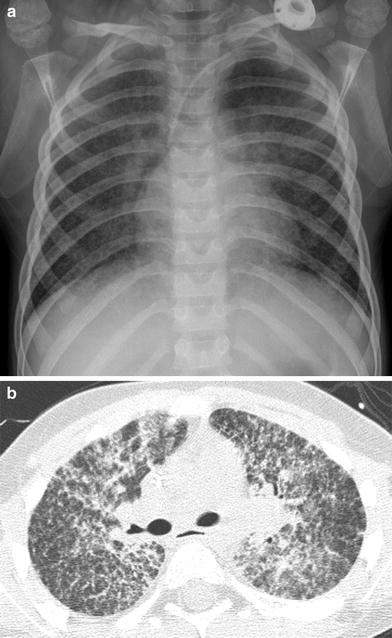
Fig. 6
Atypical mycobacterial infection. An axial chest CT image in a 14-year old with a history of large cell lymphoma and bone marrow transplantation demonstrates extensive “tree-in-bud” opacities of the left lower lobe
Fig. 7
Invasive aspergillosis. An axial chest CT image (a) in a febrile neutropenic 12-year old with acute lymphoblastic leukemia undergoing induction chemotherapy shows multiple pulmonary nodules with a ground-glass “halo” in the right lung. An axial chest CT image (b) obtained 19 days later after neutrophil recovery shows cavitation of the anterior right lung lesion. An axial CT chest image (c) obtained a further 2 weeks later reveals development of a mural nodule in the cavitary lesion that was due to cytomegalovirus superinfection
Fig. 8
Pneumocystis jiroveci pneumonia (PCP). A frontal CXR (a) and an axial chest CT image (b) from a 5-year old with acute lymphoblastic leukemia undergoing maintenance chemotherapy show diffuse reticulonodular opacities with a central predominance
CT scanning can be overused in children with immunodeficiencies. In one study of children with primary antibody immunodeficiencies and chronic cough, there was little disease progression on CT over 3 years and the authors suggested that annual surveillance may be more than is necessary (Rusconi et al. 2003). While MRI currently lags CT in spatial resolution, MRI has been shown to reliably identify pneumonia in immunocompromised patients when compared to CT, and is more sensitive for necrotizing pneumonia than CT (Leutner et al. 2000).
Due to the large number of types and varied phenotypic expressions of PID, only the disorders that are the most common or have the most characteristic imaging manifestations will be further discussed in this chapter.
4.1 Predominantly Antibody Deficiencies
The predominantly antibody deficiencies result from a decreased ability to produce immunoglobulins from B-cells and, in aggregate, account for more than half of all cases of PID (Buckley 2004). Genetic defects resulting in impaired immunoglobulin formation have been identified in multiple discrete steps in B-cell development (Cunningham-Rundles and Ponda 2005). Antibody deficiencies predispose to recurrent respiratory tract infection with encapsulated bacteria such as Streptococcus and Haemophilus influenzae, leading to bronchiectasis (Buckley 2004). Despite the presence of chronic respiratory symptoms, delay in diagnosis is common and can result in progressive lung damage with development of respiratory insufficiency and cor pulmonale (Wood et al. 2007).
4.1.1 IgA Deficiency
IgA deficiency is the most common PID and is noted in as many as 1 in 333 blood donors (Cunningham-Rundles 2001). IgA is secreted onto epithelial surfaces, and is present in smaller amounts in serum as well. Most individuals with IgA deficiency are asymptomatic. However, an increased propensity for infections of the respiratory, gastrointestinal, and genitourinary tracts is seen in some affected individuals. Pyogenic sinopulmonary infections are the most common (Cunningham-Rundles 2001), and the presence of IgA and IgG subclass antibody deficiencies is associated with greater pulmonary damage in children with recurrent respiratory tract infections (Ozkan et al. 2005).
4.1.2 X-Linked Agammaglobulinemia
X-linked agammaglobulinemia (XLA) is responsible for approximately 85 % of cases of childhood agammaglobulinemia (Plebani et al. 2002). There may be a family history of an affected brother, uncle, or male cousin. XLA most often results from a mutation in the X-linked gene encoding the Bruton tyrosine kinase. Deficiency of this kinase prevents precursor cells from maturing into circulating B-cells and plasma cells, resulting in impaired production of immunoglobulins of all isotypes (Buckley 2004). Infants with XLA typically present after the first several months of life coinciding the waning of maternally transmitted IgG (Fried and Bonilla 2009). Without IgG therapy, recurrent bacterial infections occur (Fig. 9). Bronchiectasis due to recurring infections usually affects the lower lungs. Similar involvement of both the right and left lungs may help differentiate these patients from those with bronchiectasis related to aspiration (Curtin et al. 1991). Pulmonary insufficiency is a common cause of death. Streptococcus, H. influenzae and Mycoplasma are the most typical cause of infections. There is an increased rate of hepatitis and enterovirus infections, but other viral infections are usually handled normally (Winkelstein et al. 2006). Pneumocystis and other fungal infections are rare. The adenoids, tonsils, and lymph nodes are typically small, while the thymus is usually normal in size. A lateral airway radiograph demonstrating diminutive adenoid tissue is the absence of a history of adenoidectomy can be very helpful in suggesting this diagnosis (Buckley 2004) (Fig. 9).
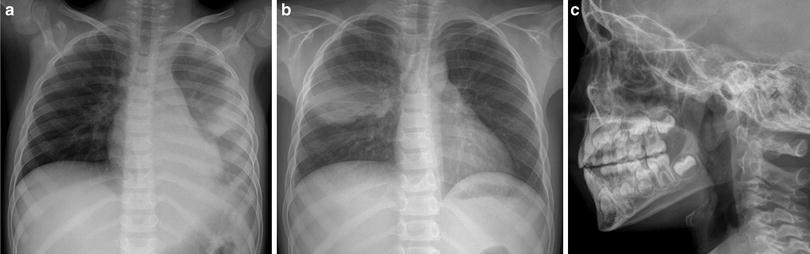
Fig. 9
X-linked agammaglobulinemia (XLA). A frontal CXR (a) from a 7-year-old boy with XLA depicts patchy air space opacity due to pneumococcal pneumonia. A frontal CXR (b) obtained 3 months later shows clearance of the previous pneumonia, but a new pneumonia of the right mid lung. A lateral upper airway radiograph (c) demonstrates diminutive adenoidal tissue
4.1.3 Common Variable Immunodeficiency Disorder
Common variable immunodeficiency (CVID) is characterized by a failure of terminal differentiation of B-cells to plasma cells, resulting in hypogammaglobulinemia, even though B-cell number may be normal. CVID is usually less severe than agammaglobulinemia and diagnostic delay until the second decade or later is common. The infectious manifestations are otherwise similar to XLA. Unlike the case in XLA, the tonsils, adenoids, and lymph nodes are not small, and may be enlarged. Splenomegaly is seen in about 25 % of cases. Also unlike XLA, CVID is associated with an increased risk for autoimmune cytopenia, lymphoproliferative disease, lymphoma and gastric cancer. The benign lymphoproliferative disease associated with CVID can have a waxing and waning course. The associated lymphadenopathy can be difficult to discern from lymphoma (Cunningham-Rundles 2012) (Fig. 10). The word variable in this disorder refers to the variability of disease severity between patients, and not to temporal variability of disease in a single patient. There are two main phenotypes, one in which infections are characteristic and another in which inflammatory or hematologic complications are prevalent (Cunningham-Rundles 2012).
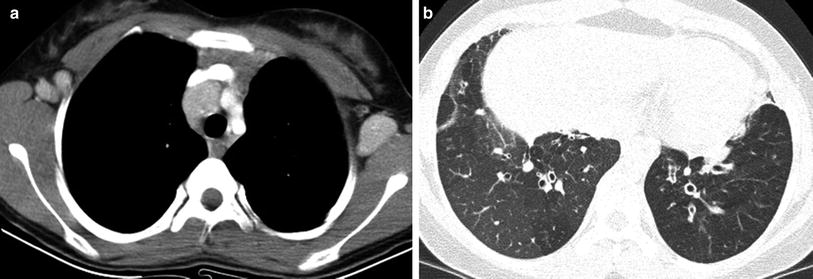
Fig. 10
Common variable immunodeficiency (CVID). An axial contrast-enhanced chest CT image (a) in a 12-year old demonstrates mediastinal and axillary lymphadenopathy representing CVID-associated benign lymphoproliferative disease that can simulate lymphoma. An axial chest CT image (b) from the same patient 2 years later shows lower lobe bronchiectasis, bronchial wall thickening, and mosaic lung attenuation from air trapping, resembling changes of cystic fibrosis
Approximately three-fourths of CVID patients develop a lung disease with air trapping, bronchiectasis, bronchial wall thickening, and endobronchial mucus plugging closely resembling cystic fibrosis (CF) (Fig. 10). Although this CF-like lung disease is associated with obstructive PFTs, it can be asymptomatic and progress silently. The degree of bronchiectasis at presentation inversely correlates with survival, and the disease can be monitored with a CF-like CT scoring system, although there is no consensus on the optimal surveillance strategy (Touw et al. 2010).
Patients with CVID can also develop granulomatous lymphocytic-interstitial lung disease (GLILD) characterized by noncaseating granulomatous and lymphoproliferative histologic patterns. Findings on chest imaging can include pulmonary nodules, consolidation, and ground-glass or reticular opacities (Fig. 11). GLILD can precede the diagnosis of CVID and is associated with restrictive PFTs, a high prevalence of autoimmune disorders and decreased survival (Park and Levinson 2010).
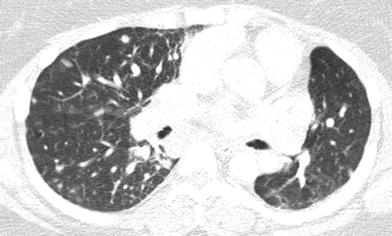
Fig. 11
Granulomatous lymphocytic-interstitial lung disease (GL-ILD). An axial chest CT image of a 21-year old with CVID shows multiple pulmonary nodules
4.1.4 Hyper-IgM Syndrome
Children affected by Hyper-IgM syndrome are identified by an elevated or normal IgM level and decreased IgG and IgA levels in serum. There is a normal number of circulating lymphocytes but a defect in signaling between T-cells and B-cells that prevents the switch from producing IgM to IgG or IgA. Most commonly, it is related to an X-linked mutation of the CD40L gene encoding a T-cell surface ligand. Presentation is similar to agammaglobulinemia, with recurrent respiratory and gastrointestinal tract infections by encapsulated bacteria, viruses, fungi and parasites. There is a particular susceptibility for P. jirovecii pneumonia (PCP) (Fig. 12). There is also a proclivity for Cryptosporidium-related diarrhea and sclerosing cholangitis, as well as an increased risk of hepatocellular carcinoma (Winkelstein et al. 2003).
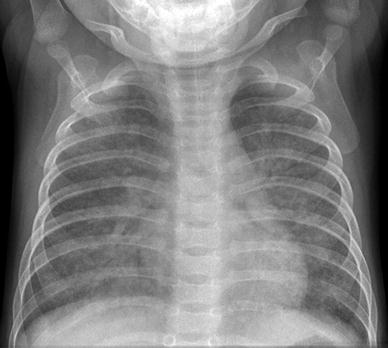
Fig. 12
X-linked hyper-IgM syndrome. A frontal CXR of a 6-year-old boy with tachypnea and hypoxemia due to Pneumocystis jiroveci pneumonia demonstrates diffuse bilateral nodular pulmonary opacities with central confluence of the opacities
4.2 Combined T-Cell and B-Cell Immunodeficiencies
T-cells function in the initial response to an antigen and in limiting the potentially harmful immune response. Because antibody production by B-cells is regulated by T-cells, T-cell immunodeficiencies may be accompanied by impaired antibody production. Children with combined T-cell and B-cell immunodeficiencies are susceptible to the same pathogens that afflict those with antibody deficiencies, as well as a variety of opportunistic infections including Mycobacteria, viruses, Pneumocystis and other fungi (Buckley 2004).
4.2.1 Severe Combined Immunodeficiency
Absence of both T-cell and B-cell function results in severe combined immunodeficiency (SCID), the most severe of the primary immunodeficiencies. A number of different genetic defects can cause SCID, including autosomal recessive and X-linked forms (Chan and Puck 2005). Most cases present between 2 and 6 months of age as protection from maternal antibodies wanes. Recurrent, persistent, severe or opportunistic respiratory infections beginning in early infancy along with thrush, dermatitis, chronic diarrhea or failure to thrive should raise suspicion of SCID. Without HSCT, gene therapy or enzyme replacement therapy, SICD is almost uniformly fatal in the first two years of life, making early diagnosis critical to prevent death from overwhelming infection (Griffith et al. 2009).
The primary role of radiology of the thorax in these children is to evaluate infections that are the main cause of mortality. While SCID patients have a diminutive thymus, physiologic thymic involution in response to stress in normal infants limits the use of this finding for suggesting a diagnosis of SCID (Manson et al. 2000) (Fig. 13). The adenosine deaminase (ADA)-deficient form of SCID is of particular note to radiologists because it is associated with a characteristic chondro-osseous dysplasia that manifests with costochondral junction cupping and splaying, concavity of the lateral margins of the vertebral transverse processes and medial margins of the posterior ribs, and thick growth arrest lines (Chakravarti et al. 1991) (Fig. 13).
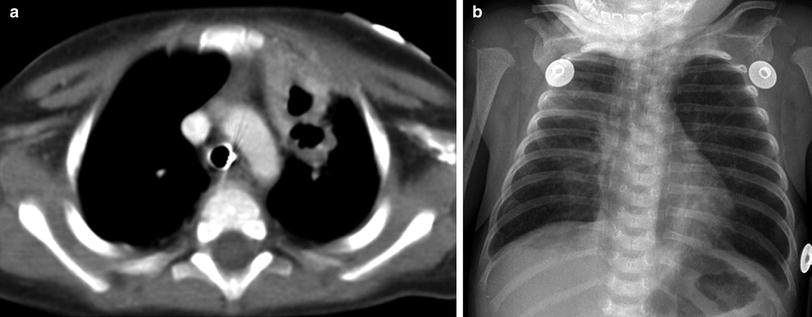
Fig. 13
Severe combined immunodeficiency (SCID). An axial contrast-enhanced chest CT image (a) in a 6-year old with SCID demonstrates a paucity of thymic tissue in the anterior mediastinum, along with cavitary lesions in the anterior left upper lobe related to Nocardia infection. A frontal CXR (b) in a 2-month old with adenosine deaminase (ADA)-deficient SCID shows cupping and splaying of the anterior rib costochondral junctions, a narrow superior mediastinum related to a diminutive thymus, and patchy bilateral pneumonia
4.2.2 Combined Immunodeficiency
While the susceptibility to infections may be similar to SCID, there is no lymphopenia in combined immunodeficiency (CID). Some forms of CID have distinctive clinical features. For example, signal transducer and activator of transcription 5B (STAT5b)-deficiency manifest with severe growth hormone-resistant growth failure beginning after birth, recurrent infection, dysmorphic features, atopic disease, autoimmune diatheses, and chronic lung disease beginning in infancy or childhood. The chronic lung disease is typically in the form of LIP (Fig. 14), and can lead to death from respiratory failure (Nadeau et al. 2011).
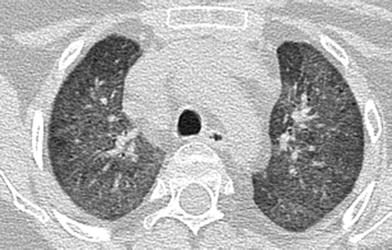
Fig. 14
Combined immunodeficiency (CID). An axial CT chest image from a 12-year old with STAT5B mutation-related CID and chronic respiratory symptoms depicts extensive bilateral ground-glass pulmonary opacities and scattered tiny cysts related to lymphocytic interstitial pneumonitis (LIP)
4.3 Well-defined Syndromes with Immunodeficiency
4.3.1 DiGeorge Syndrome
DiGeorge syndrome (DGS), also known as velocardiofacial syndrome, is a contiguous gene syndrome due to an inherited autosomal dominant or de novo chromosome 22q11.2 deletion. DGS is characterized by maldevelopment of the third and fourth pharyngeal pouches resulting in distinctive facial features (hypertelorism, saddle nose, short philtrum, low-set ears, cleft palate), conotruncal malformations (including tetralogy of Fallot, interrupted aortic arch, or truncus arteriosus), thymic hypoplasia (leading to immunodeficiency from T-cell lymphopenia and dysfunction) and parathyroid hypoplasia (leading to hypocalcemia from hypoparathyroidism) (Demczuk and Aurius 1995). Presentation in the neonatal period is more often due to hypocalcemia-induced seizures than to immunodeficiency.
DGS may be partial or complete. Partial DGS is associated with mild defects in T-cell numbers and highly variable degrees of immunodeficiency. Patients with partial DGS may experience recurrent sinopulmonary infections. Complete DGS is a form of SCID and is associated with severe T-cell deficiency. Patients with complete DGS are susceptible to opportunistic infections and to graft-versus-host disease from nonirradiated blood products (Jawad et al. 2001).
Thoracic imaging findings of DGS may include an abnormal cardiac silhouette related to a conotruncal malformation, a narrow mediastinum related to a small thymus, and pneumonia (Fig. 15). Additional findings may include tracheobronchomalacia and anomalies of the ribs or vertebrae (Ryan et al. 1997).
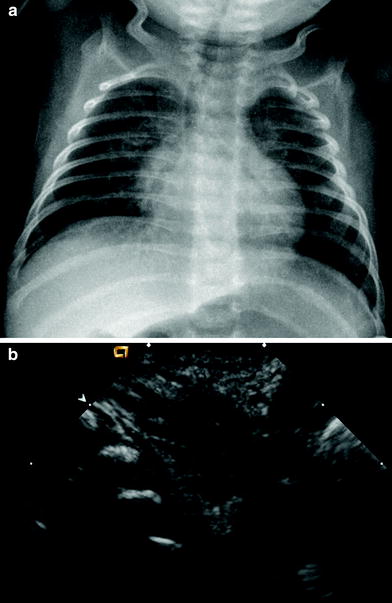
Fig. 15
DiGeorge syndrome (DGS). The thymic size is unable to be reliably assessed on the frontal CXR (a) of a 5-week-old boy with hypocalcemic seizures from parathyroid hypoplasia. A longitudinal chest US image (b) reveals a hypoplastic thymus
4.3.2 Wiskott-Aldrich Syndrome
X-linked gene mutations resulting in aberrant structure of the Wiscott-Aldrich syndrome (WAS) protein are responsible for this immunodeficiency. In addition to a role in immune defense, the WAS protein functions in a pathway that protects against autoimmune disease (Notarangelo et al. 2008). Clinical manifestations of WAS include recurrent infections, eczema, elevated IgA, autoimmune disease (vasculitis, colitis, glomerulonephritis), malignancy (especially non-Hodgkin lymphoma associated with Ebstein-Barr virus), and thrombocytopenia with small platelets. Subtypes exist, with phenotypes ranging from mild intermittent thrombocytopenia to the complete syndrome (Massaad et al. 2013). The association of WAS with abnormal blood clotting that leads to bleeding in the first month of life is unique among the forms of PID. Pulmonary infections with encapsulated Streptococcus pneumoniae and Pneumocystis are common in affected children. Herpes virus infections also occur (Buckley 2004) (Fig. 16). The only curative treatment is HSCT or gene therapy (Aiuti et al. 2013).
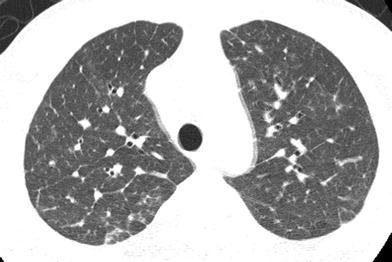
Fig. 16
Wiskott-Aldrich syndrome (WAS). An axial chest CT image from a 14-year old with a history of eczema and bloody diarrhea in infancy shows septal thickening related to herpes interstitial pneumonitis
4.3.3 Hyper-IgE syndrome
Hyper-IgE syndrome (HIES) is characterized by a reduction in CD4+ T-cells, eosinophilia and elevated IgE levels. The autosomal dominant form of HIES, also known as Job syndrome, is due to STAT3 gene mutations (Heimall et al. 2010). Job syndrome is characterized by distinctive facial features (broad nasal bridge, high palate, delayed shedding of primary teeth), eczematous dermatitis beginning in infancy, hyper-extensible joints, osteoporosis, fractures, scoliosis, and recurrent Staphylococcus aureus and Candida infections. Skin infections often begin in infancy and can manifest as “cold” abscesses that lack classical symptoms and signs of inflammation. Sinopulmonary infections are common and can occur in patients who remain afebrile, likely due to the impaired inflammatory response. Thoracic findings include recurrent pneumonia, bronchiectasis, postinfectious pneumatoceles, lymphadenopathy, and esophagitis. Pathologic fractures incurred after mild trauma can mimic child abuse (Woellner et al. 2010; Chandesris et al. 2012) (Fig. 17).
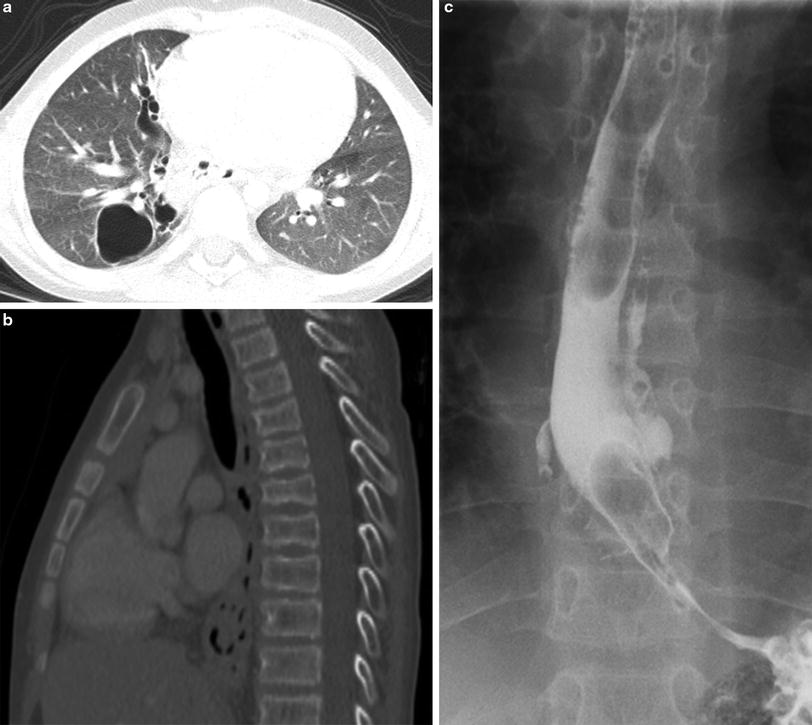
Fig. 17
Hyper-IgE syndrome (HIES). An axial chest CT image (a) from an 11-year old with HIES shows pneumatoceles in the right lung related to previous Staphylococcus infections. In the same patient, several thoracic vertebral body compression fracture deformities related to osteoporosis are revealed on a sagittal chest CT image reconstructed with bone windows (b). An esophagram (c) on the same patient demonstrates intramural tracking of contrast related to deep esophageal ulcers from Candida esophagitis
4.3.4 Dyskeratosis Congenita
Dyskeratosis congenita (DKC) is a progressive multisystem disorder characterized by the clinical triad of nail dystrophy, lacy reticular skin pigmentation, and oral leukoplakia. Additional features may include developmental delay, short stature, cerebellar hypoplasia, esophageal stenosis, urethral stenosis, osteopenia, liver fibrosis, and pulmonary fibrosis. DKC is most often attributable to mutations in genes involved in telomere maintenance, and inheritance may follow an X-linked, autosomal dominant or autosomal recessive pattern. Patients with DKC are at very high risk of aplastic anemia, myelodysplastic syndrome, leukemia, and squamous cell carcinoma. The bone marrow failure does not respond to immunosuppressive therapy (Savage and Alter 2009). Pulmonary fibrosis is a serious complication and accounts for more than 15 % of deaths in DKC. The pulmonary fibrosis occurs earlier in DKC patients who undergo HSCT and is rapidly progressive with a median survival of less than 2 years after onset of pulmonary symptoms. The presenting features of DKC-related pulmonary fibrosis include persistent dry cough, progressive dyspnea, restrictive PFTs, markedly reduced DLCO, and patchy or diffuse interstitial pulmonary opacities on CXR or CT (Giri et al. 2011) (Fig. 18).
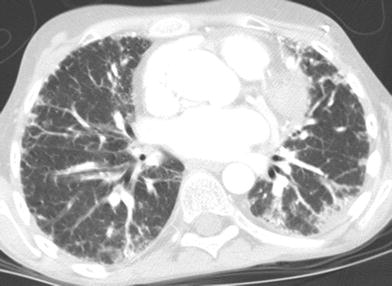
Fig. 18
Dyskeratosis congenita (DKC). An axial chest CT image of a 12-year old shows diffuse bilateral linear and patchy pulmonary opacities, predominantly in a subpleural distribution, representing DKC-related pulmonary fibrosis
4.3.5 Ataxia-Telangiectasia
Ataxia-telangiectasia (AT) is attributable to autosomal recessive mutations in the ATM gene that encodes a DNA damage response protein. Patients with AT have aberrant T-cells which results in onset of immunodeficiency in infancy. Cerebellar ataxia manifests at toddler age, and is often misdiagnosed as cerebral palsy. Oculocutaneous telangiectasias begin to appear at 3–6 years of age, while growth retardation develops later in childhood. An elevated AFP is characteristic and AFP testing in children with persistent ataxia is advised to avoid diagnostic delay (Cabana et al. 1998). Recurrent sinopulmonary bacterial infections and noninfectious granulomatous inflammation of various tissues are typical. In children and adolescents with AT, S. aureus, H. influenzae, and S. pneumoniae are the predominant respiratory infectious agents, while opportunistic infections are not observed (Schroeder and Zielen 2013). There is extreme susceptibility to DNA damage from ionizing radiation and a high risk of cancer, especially lymphoma, leukemia, and leiomyosarcoma. Life expectancy is only 20–30 years due to the increased mortality from cancer and respiratory disease (Micol et al. 2011).
Thoracic imaging findings can include a small thymus, recurrent pneumonia, bronchiectasis, lymphadenopathy, and a chronic interstitial lung disease unique to ataxia-telangiectasia (AT-ILD). AT-ILD is characterized by lymphocytic infiltrate, bizarre atypical cells, and fibrosis resulting in interstitial and pleural thickening, restrictive PFTs, chronic cough, fever, dyspnea, and an inability to reinflate the lung after pneumothorax (Fig. 19). AT-ILD has a mean age of onset of 17.5 years and a prevalence of at least 25 % in AT patients (Schroeder et al. 2005). Progressive clinical deterioration and death usually ensue within 2 years of diagnosis of AT-ILD, unless steroid therapy is initiated within the first year of onset (McGrath-Morrow et al. 2010).
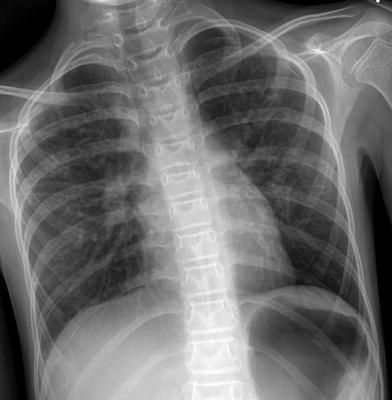
Fig. 19
Ataxia-telangiectasia (AT). A frontal CXR in a 13-year old with AT and worsening dyspnea demonstrates subtle interstitial opacities, predominantly in the mid and upper lungs, and a small left pneumothorax. These findings are manifestations of a unique interstitial lung disease (AT-ILD) associated with progressive clinical deterioration and high mortality
4.4 Congenital Defects of Phagocyte Number and/or Function
Congenital defects of phagocyte number and/or function manifest with variable susceptibility to infection and exist in neutropenic and nonneutropenic forms. The neutropenic forms can be nonsyndromic or syndromic, as in the case of Shwachman-Diamond syndrome (pancytopenia, exocrine pancreatic insufficiency, and chondrodysplasia). Nonneutropenic forms are subdivided according to the results of a dihydrorhodamine (DHR) assay. A normal DHR assay is seen in pulmonary alveolar proteinosis from alveolar macrophage dysfunction due to CSF2-receptor-alpha (GM–CSF-receptor-alpha) gene defects, and in Mendelian susceptibility to mycobacteria disease (MSMD) from an abnormal IL12-IFNgamma axis due to IFNGR1 gene defects. An abnormal DHR assay is seen in chronic granulomatous disease (CGD) (Bousfiha et al. 2013).
4.4.1 Chronic Granulomatous Disease
Due to a defect in the nicotinamide adenine dinucleotide phosphate-oxidase (NAPDH) complex, the phagocytes in children with chronic granulomatous disease (CGD) are unable to generate the superoxide burst necessary for killing ingested micro-organisms. CGD occurs in both X-linked and autosomal recessive forms, with the former being more common and severe.
Symptoms or signs of CGD often develop in the first 2 years of life. Presentation and ongoing complications are related to granuloma and abscess formation. These manifestations are likely due to ongoing inflammatory response to viable microorganisms. Histopathologic studies have shown that the granulomas in these patients may contain relatively few organisms and that the predominant portion of the granulomas results from the exuberant inflammatory response (Moskaluk et al. 1994).
Catalase-positive S. aureus is the most commonly cultured organism in the granulomas. Burkholderia and Serratia are also frequently isolated, while Streptococcus is relatively rare. Fungi and atypical mycobacteria are other causes of infection. Children with CGD are at high risk for invasive aspergillosis, which is the most common cause of mortality (Tabone 2003).
The lungs are the most common site of involvement, with pneumonia occurring in 80 % of patients (Mahdaviani et al. 2013). Lung findings on CT vary depending on the infectious organism and disease chronicity, and may include consolidation, ground-glass opacities, centrilobular opacities, nodules, bronchiectasis, septal thickening, cysts, and parenchymal distortion (Towbin and Chaves 2010). Chronic lung involvement may lead to pulmonary fibrosis, honeycombing, pulmonary hypertension, and development of systemic blood supply to areas of pulmonary infection (pseudo-sequestration) (Matsuzono et al. 1995). Extrapulmonary thoracic manifestations include suppurative lymphadenopathy, pleuritis, empyema, and rib or vertebral osteomyelitis (Khanna et al. 2005). Infections involving both the lungs and the chest wall are highly suggestive of the diagnosis of CGD (Fig. 20). Fluorodeoxyglucose-positron emission tomography (FDG–PET) is more reliable than CT for distinguishing active disease from quiescent disease or scarring (Fig. 21), an important distinction for evaluating therapy response, identifying sites for biopsy or surgical debridement, and planning for HSCT (Gungor et al. 2001) (Fig. 21).
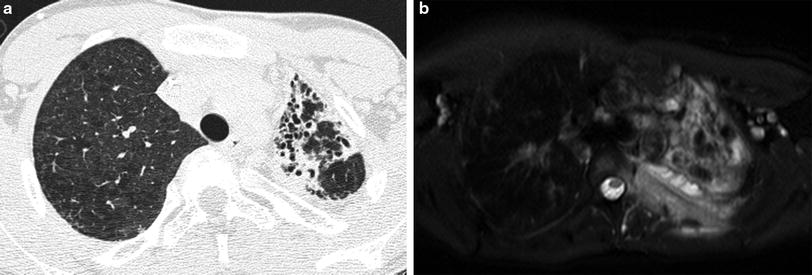
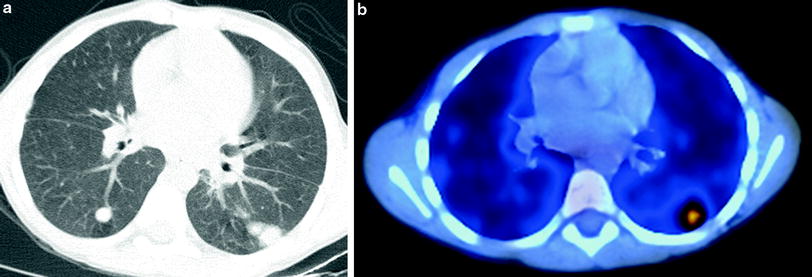
Fig. 20
Chronic granulomatous disease (CGD). An axial chest CT image (a) from a 13-year old shows left lung volume loss and traction bronchiectasis from fibrosis related to CGD. An axial T2-weighted fat saturated chest MR image (b) in the same patient at 19 years of age reveals involvement of the left pleura, ribs, and chest wall soft tissues by invasive Aspergillus
Fig. 21
Chronic granulomatous disease (CGD). An axial chest CT image (a) in a 6-year old undergoing evaluation for bone marrow transplantation for CGD shows multiple bilateral pulmonary nodules. An axial fused FDG–PET/CT image (b) reveals increased FDG uptake within only one of the nodules located in the posterior left lung. Subsequent targeted biopsy of the nodule allowed identification of active fungal infection with Cladophialophora bantiana that was undiagnosed by other means
4.5 Acquired Immunodeficiency
4.5.1 Acquired Immunodeficiency Syndrome
In the Pediatric Pulmonary and Cardiovascular Complications of Vertically Transmitted HIV (P2C2) Study from the 1990s, pulmonary infection was the most common cause of death, especially in infants and young children (Langston et al. 2001). Since then, there has been a marked decrease in opportunistic pulmonary infections and deaths related to acquired immunodeficiency syndrome (AIDS) in developed countries as a result of the implementation of highly active antiretroviral therapy (HAART) (Gona et al. 2006). Even in developed countries, though, vigilance for opportunistic infections remains important due to noncompliance and drug resistance.
Although HAART has reduced rates of opportunistic infection, bacterial pneumonia still tends to be more severe in HIV-infected children than in HIV-uninfected children related to a higher incidence of abscess and empyema. Streptococcus is the most common pathogen, and manifests as lobar or multilobar patchy consolidation. Mycobacterial infection is more likely to be multilobar or miliary, and is associated with necrotic lymphadenopathy. Pneumonias from respiratory syncytial virus (RSV), cytomegalovirus (CMV), parainfluenza, influenza, adenovirus, or human metapneumovirus infections have diverse radiographic appearances that can be indistinguishable from bacterial pneumonias. A patient age of less than 6 months, a respiratory rate >59 breaths/minute, an O2 saturation <93 %, and an absence of vomiting are clinical features that suggest P. jirovecii pneumonia (PCP), which classically manifests with bilateral diffuse or patchy ground-glass or reticulonodular opacities with perihilar to peripheral progression, although this appearance overlaps with mycobacterial and viral pneumonia. PCP can also be associated with pulmonary cysts that rupture, leading to pneumothorax or pneumomediastinum (George et al. 2009).
Lymphocytic interstitial pneumonitis (LIP) is a form of pulmonary lymphoid hyperplasia characterized by infiltration of the interstitium by lymphocytes and plasma cells and classically presenting in HIV-infected children older than 2 years of age with chronic or recurrent cough and mild hypoxemia (Theron et al. 2009). The 1987 CDC criteria for a presumptive diagnosis stipulate the persistence of diffuse, symmetrical, reticulonodular or nodular pulmonary opacities for at least 2 months without an identifiable pathogen, or a response to antibiotic therapy (Pitcher et al. 2010). CT reveals nodules in a subpleural, septal, centrilobular, or peribronchial distribution, and ground-glass opacities with or without lymphadenopathy (Becciolini et al. 2001) (Fig. 22). Bronchiectasis and cystic dilated air spaces have also been described (Pitcher et al. 2010). Resolution of LIP in children can be due to response to HAART or corticosteroid therapy, or to immunologic deterioration and progression of HIV disease (Theron et al. 2009).
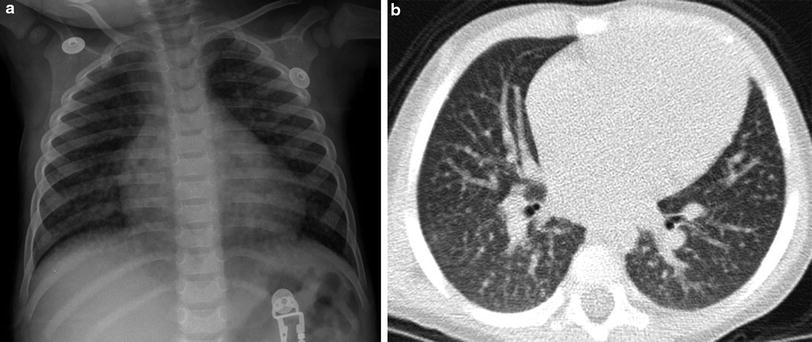
Fig. 22
Lymphocytic interstitial pneumonitis (LIP). A frontal CXR (a) in a 16-month-old HIV+ patient shows numerous tiny pulmonary nodules. No infectious pathogen was identified, and the nodules persisted for several months, as demonstrated on a chest CT (b) obtained at 21 months of age
The immune reconstitution inflammatory syndrome (IRIS) is an exaggerated inflammatory response by the reconstituted immune system occurring 1–6 months after HAART initiation, resulting in apparent clinical worsening. “Unmasking” IRIS occurs in response to a previously unrecognized underlying infection, while “paradoxical” IRIS occurs in response to a previously treated infection. IRIS is most frequently observed in response to mycobacterial, herpes virus, JC virus, PCP, and cryptococcal infections, and can appear as new or worsening lymphadenopathy, pulmonary nodules, or pulmonary consolidation on imaging. IRIS must be distinguished from noncompliance, drug resistance, or newly acquired opportunistic infection, and is treated by NSAIDs, steroids, and discontinuation of HAART (Theron et al. 2009).
A high incidence of swallowing dysfunction and gastroesophageal reflux predisposes to aspiration pneumonia in HIV-infected children. These children may also suffer from odynophagia and dysphagia related to esophagitis caused by Candida or CMV infection. HIV-related cardiomyopathy, antiviral therapy cardiotoxicity, accelerated atherosclerosis and chronic anemia can lead to cardiac failure and pulmonary edema that is misattributed to primary pulmonary infection (Pitcher et al. 2009). In addition, HIV infection is associated with an increased risk of thymic cysts and non-Hodgkin lymphoma, which are covered in the chapter entitled Imaging of the Pediatric Thymus and Thymic Disorders by Sams and Voss, as well as smooth muscle tumors, which are covered in the chapter entitled Pulmonary and Extra-Thymic Mediastinal Tumors by Lyons, Guillerman and McHugh.
4.5.2 Acquired Neutropenia and Invasive Fungal Disease
Neutropenia is common in children undergoing chemotherapy for cancer or allogeneic HSCT, and confers a high risk of invasive fungal disease (IFD) by mold (most frequently Aspergillus) or yeast (most frequently Candida). The lungs are the most common site of deep organ involvement by IFD. In a neutropenic patient with fever or respiratory symptoms or signs, the traditional approach has been to obtain a CXR, give early empiric broad-spectrum antibacterials, and add empiric antifungals for suspected invasive fungal disease (IFD) if the fever or respiratory symptoms or signs persist >96 h. In the presence of a deficient immune response, findings of infection are often absent or subtle and nonspecific on CXR, so that chest CT is preferred. However, even the yield of chest CT is low in this setting. In a study of 52 pediatric oncology patients with persistent febrile neutropenia, only 18 % of the initial (mean of 5 days of febrile neutropenia) CT scans were positive, and alterations in therapy were made in only 3 % based on the findings of initial CT. Repeat (mean of 17 days of febrile neutropenia) CT had a much higher yield, being positive in 52 % of cases and directing a change in therapy in 20 %. Respiratory symptoms or signs were present in only a minority of cases with a positive chest CT, while all patients with a concern for occult IFD had findings on chest CT, suggesting that initial chest CT should be limited to patients without localized signs or symptoms (Agrawal et al. 2011).
Rather than administering empiric antifungals in all patients at risk with persistent febrile neutropenia, a “pre-emptive” or “diagnostic-driven” approach of administering antifungals only in the setting of either microbiologic (cytology, direct microscopy, culture, ß-D-glucan or galactomannan assay of serum, or bronchoalveolar lavage fluid) or chest CT evidence of probable IFD may reduce unnecessary therapy without compromising prognosis (Castagnola et al. 2014). The European Organization for Research and Treatment of Cancer/Mycoses Study Group (EORTC-MSG) chest CT criteria for probable IFD are the presence of well-circumscribed pulmonary lesions (with or without a “halo” sign), an “air-crescent” sign, or a cavity in the appropriate clinical setting (De Pauw et al. 2008).
The “halo” sign consists of a ground-glass halo of alveolar hemorrhage around a nodular or consolidative focus of infarcting lung from fungal vascular invasion and thrombosis that occurs early in IFD. A “reversed halo” sign of focal ground-glass opacity surrounded by a crescent or ring of consolidation may also be observed with early IFD (Georgiadou et al. 2011). An “air-crescent” sign or cavitation is the result of neutrophil recovery with release of proteases that resorb necrotic tissue (McAdams et al. 1995). In the course of IFD, a “halo” sign, nodule, or consolidation is typically visible on chest CT within 5 days of fever onset (Fig. 7). Early initiation of antifungals based on detection of the “halo” sign on chest CT is associated with better response to treatment and improved survival (Greene et al. 2007). Despite effective antifungal therapy, an increase in lesion number and size may be observed for 7–10 days followed by a plateau for a few days, and should not be confused for progressive disease. The “air-crescent” sign and cavitation develop in 2–3 weeks, dependent on the timing of neutrophil recovery (Caillot et al. 2001) (Fig. 7). Refractory IFD is suggested by no decrease in lesion number or size or by lack of development of an “air-crescent” sign or cavitation after 2–3 weeks, but initial or maximal lesion size or number does not impact outcome (Horger et al. 2005). After 2–3 weeks, reappearance of a “halo” sign, development of new lesions, or an increase in wall thickness of a cavitary lesion suggests relapsed or new infection (Fig. 7). Even after clinical remission, there may be fibrotic or thin-walled cavitary remnants visible on chest imaging (Gefter et al. 1985).
The chest imaging findings of IFD are not entirely specific and can also be observed in immunocompromised children with Myobacterium, Nocardia, Pseudomonas, or herpes virus infections (Gasparetto et al. 2008). Following allogeneic HSCT, children are susceptible not only to opportunistic pulmonary infections, but also to noninfectious pulmonary disorders such as alveolar hemorrhage, pulmonary edema, drug reaction, lymphoproliferative disease, organizing pneumonia, bronchiolitis obliterans, graft-versus-host disease, and idiopathic pneumonia syndrome. These disorders can cause considerable morbidity and mortality in the post-transplant setting, and can be difficult to distinguish on the basis of imaging findings, so that correlation with clinical features, identification of risk factors and institution of preventative measures are paramount in management (Sakaguchi et al. 2012).
5 Vasculitis
Vasculitis is an inflammatory disease of the blood vessel walls. Apart from invasive aspergillosis, infectious vasculitis is uncommon in childhood. Environmental, autoimmune and genetic factors play a role in the pathogenesis of noninfectious vasculitis. Vasculitis can be categorized according to the predominant size of the affected vessels. Takayasu arteritis, a large vessel vasculitis, and Kawasaki disease, a medium vessel vasculitis, are covered in the chapters entitled Pediatric Cardiac CT by Sena and Goo and Pediatric Cardiac MRI by Krishnamurthy and Chung. Small vessel vasculitis affecting the pediatric lungs may be associated with a systemic disease (such as SLE), an immune complex deposition disease (such as anti-glomerular basement membrane disease), or diseases with anti-neutrophil cytoplasmic antibodies (ANCA). ANCA-associated vasculitides are characterized by necrotizing inflammation of small vessels (i.e., capillaries, venules, arterioles, small arteries) and encompass overlapping syndromes, including granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) (Jennette et al. 2013). ANCA directed against proteinase 3 (PR3) are predominantly associated with GPA, and ANCA directed against myeloperoxidase (MPO) are mainly associated with MPA (Millet et al. 2013).
Pediatric vasculitis affecting the lungs is often associated with diffuse pulmonary hemorrhage. This must be distinguished from nonvasculitic causes of pulmonary hemorrhage in childhood, such as idiopathic pulmonary hemosiderosis, cystic fibrosis, coagulopathy, pulmonary venoocclusive disease, and pulmonary arteriovenous malformations (Susarla and Fan 2007).
5.1 Granulomatosis with Polyangiitis
Granulomatosis with polyangiitis (GPA), formerly known as Wegener granulomatosis, is characterized by necrotizing vasculitis predominantly affecting small to medium vessels (i.e., capillaries, venules, arterioles, arteries, and veins), usually of the upper and lower respiratory tract. Necrotizing glomerulonephritis is also common. A limited form initially confined to the lungs may occur. In the acute phase, the predominant pattern of inflammation is purulent rather than granulomatous. In the chronic phase, granulomatous and nongranulomatous extravascular inflammation is common (Jennette et al. 2013). The most common finding at presentation on chest imaging is multiple pulmonary nodules and masses. The lesions may coalesce or cavitate over a period of several days or a few weeks (Fig. 23). Consolidation and ground-glass opacities may also be noted. The airways can be affected, manifesting as tracheobronchial wall thickening, bronchial stenosis or bronchiectasis (Castaner et al. 2010).
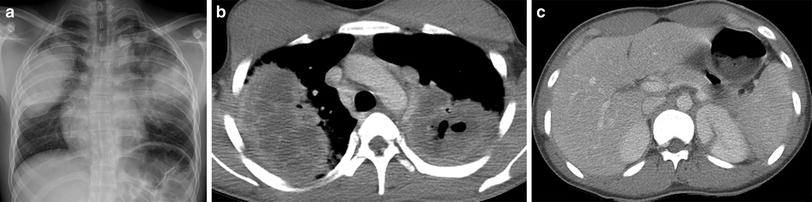
Fig. 23
Granulomatosis with polyangiitis (GPA). A frontal CXR (a) of a 17-year-old male with hemoptysis, hematuria and elevated anti-PR3 antibodies depicts large mass-like opacities of the mid to upper lungs bilaterally. An axial contrast-enhanced chest CT image (b) demonstrates low-attenuation necrosis and cavitation within the pulmonary opacities. An axial contrast-enhanced abdominal CT image (c) reveals a wedge-shaped lesion of the posteromedial aspect of the left kidney related to renal vasculitis
5.2 Microscopic Polyangiitis
Microscopic polyangiitis (MPA) is characterized by necrotizing vasculitis predominantly affecting small vessels (i.e., capillaries, venules, or arterioles). In distinction to GPA, granulomatous and extravascular inflammation is absent. Renal and lung involvement are common.
In pulmonary capillaritis, there is inflammatory disruption of the alveolar interstitial capillary network, resulting in pulmonary hemorrhage. Although most pediatric patients with pulmonary capillaritis have dyspnea and anemia on presentation, only a minority has hemoptysis, contributing to diagnostic delay (Fullmer et al. 2005). ANCA-associated pulmonary capillaritis is an underappreciated cause of pulmonary hemorrhage and likely accounts for a substantial proportion of pulmonary hemorrhage cases attributed to idiopathic pulmonary hemosiderosis (Taytard et al. 2013).
Acute hemorrhage into the pulmonary airspaces produces ill-defined pulmonary nodules, ground-glass opacities and consolidation on CT (Fig. 24). Aggregates of hemosiderin-laden macrophages from repetitive hemorrhage result in septal thickening and crazy-paving. Parenchymal and subpleural cysts and architectural distortion may be observed on CT in the chronic setting (Connolly et al. 1996; Ravenel and McAdams 2003).
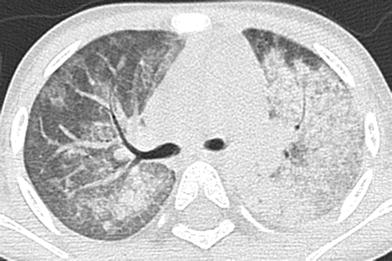
Fig. 24
Microscopic polyangiitis (MPA). An axial chest CT image of a 2-year-old girl with anemia, hypoxemia and elevated anti-MPO antibodies show patchy ground-glass opacities of the right lung and consolidative opacities of the left lung. These findings represent alveolar hemorrhage related to pulmonary capillaritis
The finding of ill-defined centrilobular opacities suggest angiocentric inflammation and hemorrhage from small vessel pulmonary vasculitis, but it is not possible to reliably differentiate capillaritis from other causes of diffuse pulmonary hemorrhage on the basis of the imaging features, and lung biopsy is often required for differentiation (Guillerman and Brody 2011). Definitive diagnosis is crucial because successful treatment of pulmonary capillaritis typically requires aggressive and prolonged immunosuppressant therapy. CT can be used to help assess response to therapy (Vece and Fan 2010).
6 Sickle Cell Disease
The sickle hemoglobinopathies are autosomal recessive genetic disorders that include the presence of hemoglobin (Hgb) S within red blood cells. A substitution of valine for glutamic acid in the hemoglobin beta chain changes the structure of hemoglobin, allowing the hemoglobin to crystallize under conditions of low oxygen, dehydration, and acidosis. When the hemoglobin crystallizes, the red blood cell becomes less flexible and assumes the namesake sickle shape. These sickle cells have a decreased life span related to hemolysis and resulting in anemia. These nondeformable cells often obstruct small capillaries where slow flow promotes the conditions that encourage further sickling in a cascade effect.
Homozygous Hgb SS produces the most severe disease. Different mutations are associated with different amounts of hemoglobin F, with higher levels of Hgb F being associated with decreased disease severity. Compound heterozygous conditions occupy a spectrum of disease severity depending on the non-S hemoglobin. The most common are hemoglobin S with hemoglobin C (Hgb SC) and hemoglobin S with β0 or β+ thalassemia. Hgb Sβ0 thalassemia is similar to Hgb SS, while Hgb Sβ+ thalassemia and Hgb SC disease will often have preserved splenic function and less risk of vaso-occlusive crisis and infection. Heterozygous hemoglobin S and hemoglobin A is the asymptomatic carrier state.
Infants with sickle cell disease usually show no abnormalities, and clinical signs and symptoms develop as fetal hemoglobin is replaced by defective adult-type hemoglobin. Hand-foot syndrome, a painful swelling of the hands and feet, may be an early presentation, in which case soft tissue swelling and periosteal new bone formation are seen radiographically (Babhulkar et al. 1995). Anemia and painful crises in other locations then develop. Decreased splenic function causes an increased susceptibility to infection.
The most common cardiovascular imaging findings in children with sickle cell disease are mild cardiomegaly and pulmonary vascular plethora related to chronic anemia. Myocardial perfusion abnormalities in children have been reported (Acar et al. 2000). Bone infarctions are common. CXR may demonstrate biconcave vertebral bodies from impression of the intervertebral discs on the adjacent endplates weakened by microinfarcts.
Lung function can be abnormal from an early age. Evidence of obstructive and restrictive pulmonary disease has been shown on PFTs in infants and young children (Koumbourlis et al. 1997; Arteta et al. 2013). It is increasingly recognized that long-term adverse changes in lung function are a major cause of morbidity and mortality, even in the absence of episodes of acute lung disease (Koumbourlis 2013). Airway hyperreactivity, which is more common in individuals with sickle cell disease, is associated with obstructive physiology (Mekontso Dessap et al. 2013) and may be an additional factor in the development of both acute and chronic lung disease (Leong et al. 1997).
The two most common acute chest complications of sickle cell disease are pneumonia and the acute chest syndrome (ACS). Functional asplenia can occur as young as 6 months of age. Children are then particularly susceptible to infections caused by bacteria with polysaccharide capsules. While S. pneumoniae sepsis was once a common cause of death in young children with sickle cell disease, mortality has decreased dramatically in recent years related to the effectiveness of newborn screening, prophylactic penicillin, and vaccination measures (Hamideh and Alvarez 2013).
The acute chest syndrome (ACS) is a major cause of morbidity and mortality in sickle cell disease. ACS a largely descriptive term applied to the clinical situation in which a patient with sickle cell disease develops a new infiltrate on CXR accompanied by chest pain, fever, and respiratory symptom (Fig. 25). ACS has been reported to account for 30 % of deaths in sickle cell patients under 10 years of age (Martin and Buonomo 1997), but the absolute mortality has decreased in the modern era of hydroxyurea therapy and early aggressive transfusion therapy. In a prospective study of 538 patients admitted for ACS, overall mortality was 3 % (Vichinsky et al. 2000). In adults, ACS-associated mortality is now primarily related to underlying pulmonary hypertension and acute cor pulmonale (Miller and Gladwin 2012). A specific cause can be identified in ACS in 40–70 % of cases. Etiologies include infection, pulmonary infarction, and fat embolism from vasoocclusive crises involving bone marrow (Vichinsky et al. 2000). Rib infarctions have also been identified in ACS (Gelfand et al. 1993) (Fig. 25). Asthma is associated with an increased risk of ACS in children (Boyd et al. 2006), and episodes of ACS predispose children to increased airway resistance (Sylvester et al. 2006). Repetitive episodes of ACS also lead to lung fibrosis, predominantly at the lung bases, contributing to chronic restrictive lung disease (Miller and Gladwin 2012).