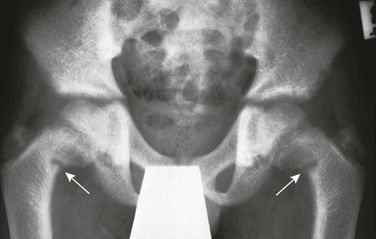
Chapter 140 In the process of bone formation, osteoblasts produce organic bone matrix (osteoid), which then must be mineralized with deposition of hydroxyapatite crystals.1–4 Mineralization of cartilage and osteoid requires sufficient levels of circulating calcium and phosphate as reflected in an adequate calcium x phosphate product. Mineralization also requires alkaline phosphatase to hydrolyze pyrophosphate, which otherwise would inhibit crystal formation.5 Hence deficient mineralization of cartilage and osteoid may be due to either an insufficient calcium x phosphate product as in rickets and osteomalacia or alkaline phosphatase deficiency as in hypophosphatasia. Rickets is a complex disorder of the growth plate that involves not only deficient mineralization of cartilage and osteoid but also disruption of endochondral ossification, leading to the accumulation of excessive cartilage. This phenomenon results from failure of hypertrophic chondrocytes to undergo normal apoptosis, which is now believed to be caused by hypophosphatemia as the common metabolic pathway of all forms of rickets (in calcipenic rickets, hypophosphatemia results from secondary hyperparathyroidism).6 Osteomalacia is a pure disorder of insufficient mineralization of osteoid at sites other than the physes, hence at either sites of bone turnover or intramembranous bone formation. Forms of rickets due to insufficient calcium are mostly a result of abnormalities of vitamin D, whose major function is maintenance of a sufficient calcium x phosphate product. Vitamin D may be synthesized in the skin from 7-dehydrocholesterol upon exposure to sunlight containing ultraviolet B radiation (290 to 315 nm) or it may be provided in the diet, although natural dietary sources of vitamin D are quite limited. Vitamin D then undergoes 25-hydroxylation in the liver followed by highly regulated 1-α-hydroxylation in the kidney to produce 1,25(OH)2-vitamin D, which is the active form of vitamin D (calcitriol). The most important biological function of calcitriol is facilitation of gastrointestinal absorption of calcium by inducing transcription of the gene for calcium-binding protein. Calcitriol synthesis by 1-α-hydroxylase is promoted by parathyroid hormone (PTH) in response to hypocalcemia. Rickets also may result from a variety of disorders that cause hypophosphatemia, in most instances from renal tubular phosphate wasting. Renal tubular phosphate wasting or retention is regulated by PTH and by a variety of other factors, most of which are produced in bone by osteocytes.7–12 The most important of these is fibroblast growth factor 23 (FGF23), which causes renal tubular phosphate wasting by downregulating the sodium-phosphate cotransporter that facilitates phosphate reabsorption. Additionally, FGF23 downregulates 1-α-hydroxylase, leading to diminished calcitriol with excessive FGF23 signaling. Similarly, insufficient FGF23 signaling increases calcitriol. Etiology: Both inadequate exposure to sunlight and insufficient dietary intake of vitamin D must be present for rickets to occur.1–3,13–16 The first and most severe epidemic of rickets occurred during the industrial revolution as a result of urbanization and diminished exposure to sunlight from smog and the practice of staying indoors. This epidemic largely ended with food fortification after the discovery of vitamin D synthesis. However, rickets then became more prevalent in the United States during the mid 1990s with increased breast feeding in the African American and Hispanic populations, because breast milk provides relatively little vitamin D. Less commonly, nutritional rickets may result from insufficient calcium intake.17 Clinical manifestations of rickets include failure to thrive, short stature, bowing deformities, and predisposition to fractures. In addition, prominence of anterior rib ends causing a “rachitic rosary” and craniotabes (i.e., a ping-pong ball deformity of the skull) may be seen. Weakness is a clinical feature of rickets, correlating with the presence of vitamin D receptors in skeletal muscle.18 Imaging: The manifestations of rickets are most pronounced with rapid bone growth with mineralization that is unable to keep pace with new bone formation. As a result the features of rickets are most pronounced in regions of greatest bone growth, particularly the distal radius and ulna, distal femur, proximal tibia, proximal humerus, and anterior rib ends. An insufficient calcium x phosphate product causes decreased mineralization of the zone of provisional calcification and lack of normal chondrocytic terminal differentiation. As a result the initial radiographic finding is rarefaction of the normally sharply defined zone of provisional calcification on the metaphyseal side of the growth plate so that metaphyseal bone fades gradually into the lucent physeal and epiphyseal cartilage (Fig. 140-1 and e-Fig. 140-2). Also seen is loss of definition of the Laval-Jeantet collar, a short cylindrical segment of the metaphysis adjacent to the growth plate that is an indicator of the most recently formed bone in young infants.19 Deficient chondrocyte terminal differentiation and apoptosis causes accumulation of disorganized cartilage in the metaphysis in addition to nonmineralized osteoid, leading to widening of the distance between the epiphysis and metaphysis, metaphyseal fraying, and metaphyseal concavity (cupping). Metaphyseal concavity varies by site, being most pronounced in the distal forearm bones (see Fig. 140-1). However, distal ulnar metaphyseal concavity with no other abnormality should be recognized as a normal finding. Metaphyseal findings that may be recognized on chest radiographs include involvement of the proximal humeral metaphyses and rib ends, producing the rachitic rosary (Fig. 140-3), although this term more properly refers to the bulbous rib ends on physical examination. The classic metaphyseal findings of rickets are best illustrated by comparison of active and healed or healing phases (see Fig. 140-1 and e-Fig 140-2). Similar but less pronounced findings may be seen in the slower growing epiphyses and small bones with loss of definition of the zone of provisional calcification surrounding the ossification centers.19 Figure 140-1 Vitamin D deficiency rickets. Figure 140-3 Anteroposterior (A) and lateral (B) chest radiograph views in a 5-month-old child with rickets from biliary atresia. e-Figure 140-2 A less severe case of Vitamin D deficiency rickets than shown in Figure 140-1. At 9½ years of age (top), distal femoral physeal widening and metaphyseal fraying is noted that resolved several months later after treatment (bottom). Long bone shaft findings lag behind those in the metaphysis. In rickets due to vitamin D abnormality, PTH rises in an attempt to restore a normal serum calcium concentration, producing secondary hyperparathyroidism (HPTH) with subperiosteal bone resorption, intracortical tunneling, and overall demineralization (Fig. 140-4). With ongoing bone remodeling, as nonmineralized osteoid replaces mineralized bone (osteomalacia), cortical demineralization and coarsening of the trabecular pattern in the shafts occurs, predisposing a person to insufficiency fractures. Paradoxically, periosteal new bone formation also may be seen in rickets even before healing, which likely is an anabolic effect of PTH (see Fig. 140-1). With healing (see Fig. 140-1, e-Fig. 140-2, and Fig. 140-4), mineral deposition in the zone of provisional calcification and restoration of chondrocyte terminal differentiation occurs. The initial radiographic finding of healing is reidentification of the zone of provisional calcification as a thin opaque line that is separated from the identifiable shaft by the intervening lucent nonmineralized cartilage and osteoid. With further healing, this metaphyseal region calcifies, which may give the false impression of rapid bone growth. Less often, newly mineralized osteoid also may be seen in the metaphyseal equivalent region of the epiphysis (e-Fig. 140-5). With healing of cortical bone, the subperiosteal osteoid becomes calcified, producing either a uniform or lamellated layer. Figure 140-4 Diaphyseal findings in a patient with severe vitamin D deficiency rickets. e-Figure 140-5 Healing rickets with mineralization of previously lucent osteoid. Treatment and Follow-up: Prevention of vitamin D deficiency is a controversial issue because of lack of consensus regarding the level of vitamin D that is considered sufficient. The 2011 Institute of Medicine report20 concluded that a serum level of 25-hydroxy-vitamin D of at least 20 ng/mL is normal, although many experts in vitamin D research argue that higher levels are needed. Accordingly, the Institute of Medicine recommends 400 IU of vitamin D daily during the first year of life and 600 IU daily thereafter, with higher amounts suggested by researchers who believe that higher serum levels are needed. Treatment guidelines for vitamin D deficiency are those of the Lawson Wilkins Pediatric Endocrine Society.21 Treatment is recommended for clinical manifestations of vitamin D deficiency or 25-hydroxy-vitamin D levels less than 15 ng/mL, beginning with higher doses than those used for prevention. With treatment, if radiographic evidence of healing is seen by 3 months, vitamin D dosage can be reduced to preventive levels. Because a major cause of treatment failure is noncompliance, intermittent high-dose vitamin D therapy also has been suggested. Malabsorption and Hepatobiliary Disease: Vitamin D malabsorption may cause rickets in disorders such as celiac disease and cystic fibrosis. Rickets with hepatobiliary disease is mostly a result of decreased intake and intestinal absorption of fat-soluble vitamins (A, D, E, and K); decreased hepatic 25-hydroxylation of vitamin D usually is not significant. Associated vitamin K deficiency in hepatobiliary disease may lead to recurrent hemarthrosis, most frequently involving the knee. Vitamin D–Dependent Rickets, Types I and II: In autosomal-recessive vitamin D–dependent rickets (VDDR) type I, a defect in the renal 1-α-hydroxylase leads to undetectable or very low calcitriol levels.22,23 Rickets in VDDR I is severe, presents in the first few months of life, and has more secondary HPTH than do other forms of rickets. VDDR I responds to physiologic doses of calcitriol. Rickets also can result from a defect in the receptor for calcitriol, causing vitamin D nonresponsiveness.23,24 The designation of VDDR type II for this disorder is inappropriate because it is resistant to all forms of vitamin D, and hence the term “calcitriol-resistant rickets” is preferred. In addition to severe rickets, approximately half of the patients have manifestations of ectodermal dysplasia. Hypophosphatemic Rickets: Hypophosphatemic forms of rickets are seen in hereditary hypophosphatemia, tumor-induced rickets and osteomalacia, and intrinsic renal tubular disease. Additionally, a major component of rickets of prematurity (i.e., metabolic bone disease of prematurity) is likely phosphate deficiency. Hereditary Hypophosphatemic Disorders: Etiology: X-linked hypophosphatemia (XLH), also known as familial vitamin D–resistant rickets, is the most common hereditary cause of hypophosphatemia, with rare autosomal-dominant and recessive variants.7,9,11,16 The discovery that the autosomal-dominant variant was due to a gain of function for FGF23 led to the recognition of FGF23 as a major phosphaturic factor. In XLH, FGF23 also is increased, although it is not clearly understood how this increase is caused by a mutation in the PHEX gene. Imaging: Lower extremity bowing usually is quite prominent in XLH (Fig. 140-6), whereas the rachitic findings of XLH are often (but not always) relatively mild. Because no hypocalcemia is present, secondary HPTH is not seen in hypophosphatemic rickets. Looser zones (a feature of osteomalacia) are present more often in XLH than in nutritional rickets, likely because of the chronicity of the mineralization disorder. Looser zones, or pseudofractures, are radiolucent lines oriented perpendicular to the cortex as a result of poor mineralization of osteoid at sites of stress, which lead to increased bone remodeling. Characteristic sites include the medial aspect of the femoral neck (Fig. 140-7), the extensor surface of the ulna, the axillary border of the scapula, and the pubic ramus, with these findings often being bilateral. In infants, craniosynostosis may complicate XLH.25,26 In older patients, XLH may be associated with enthesopathy and paravertebral ossification, similar to diffuse idiopathic skeletal hyperostosis. Figure 140-6 X-linked hypophosphatemia in a 3½-year-old girl. Treatment and Follow-up: Treatment of XLH includes oral phosphate replacement and calcitriol because phosphate replacement alone often leads to secondary HPTH. However, excessive calcitriol may lead to hypercalcemia, nephrocalcinosis, and nephrolithiasis; renal ultrasonography often is used to screen for this complication.27 Tumor-Induced Rickets and Osteomalacia (Oncologic Osteomalacia): Etiology: Hypophosphatemic rickets and osteomalacia may be caused by certain tumors and tumorlike conditions that are capable of producing FGF23, with healing of rickets after removal of the tumor.28,29 TIRO may occur at any age but is quite uncommon in children. Although a large variety of benign and malignant mesenchymal neoplasms initially were described, many of these neoplasms more recently have been recategorized pathologically as “phosphaturic mesenchymal tumor, mixed connective tissue variant.”30 Other conditions causing rickets that are considered to be within the spectrum of TIRO include neurofibromatosis, polyostotic fibrous dysplasia (with or without other features of McCune-Albright syndrome), epidermal nevus syndrome, and Gorham massive osteolysis syndrome (Fig. 140-8). Figure 140-8 “Tumor-induced” hypophosphatemic rickets in a girl aged 2 years and 4 months with Gorham massive osteolysis syndrome.
Metabolic Bone Disease
Abnormalities of Mineralization
Nutritional (Vitamin D–Deficiency) Rickets
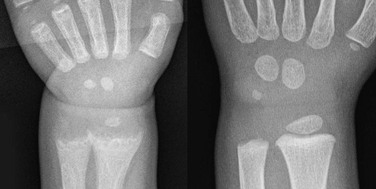
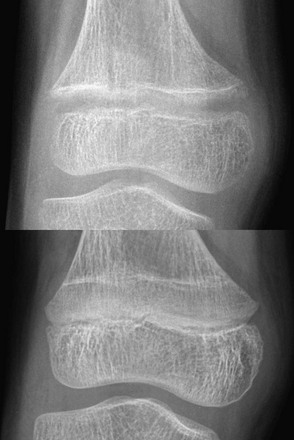
A 14-month-old child with growth failure and severe rickets who responded well to vitamin D therapy. The initial image (left) shows loss of definition of the zones of provisional calcification for the distal radial and ulnar metaphyses along with metaphyseal fraying and concavity (“cupping”) and physeal widening with an increased distance between the epiphysis and visualized portion of the metaphysis. Periosteal new bone also is present that is seen best along the metacarpals but also is present along the distal radius. With healing (right), the zone of provisional calcification is well mineralized and the other findings have resolved.
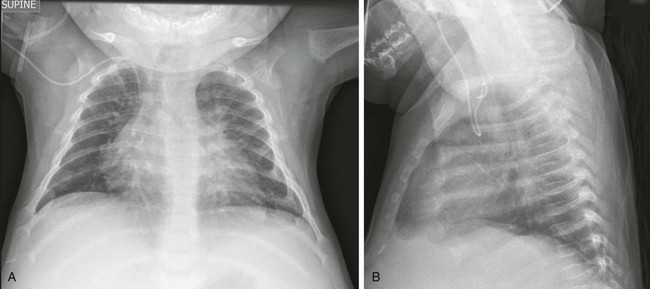
Note rickets in the proximal humeral metaphyses and anterior rib ends. The rib findings are best seen on the lateral view.
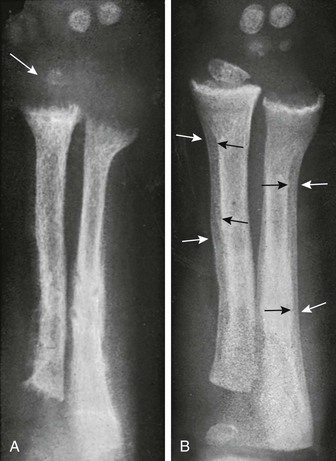
During the active phase (A), coarse demineralization and subperiosteal bone resorption are present, which are indicative of hyperparathyroidism as a result of rickets. Also note the severe rachitic findings in the metaphysis and poor mineralization of the distal radial epiphysis with loss of the zones of provisional calcification (arrow). With healing 3 months later (B), extensive periosteal new bone is seen (white arrows) with calcification of previously nonmineralized osteoid (black arrows) produced by periosteal osteoblasts.
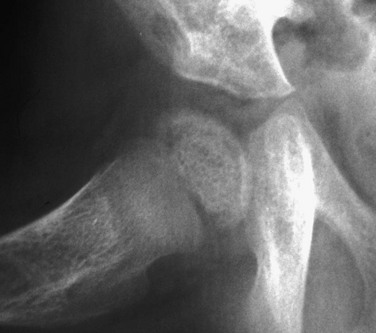
Newly mineralized bone is seen in the proximal femoral metaphysis, as well as the equivalent portion of the epiphysis.
Other Forms of Rickets
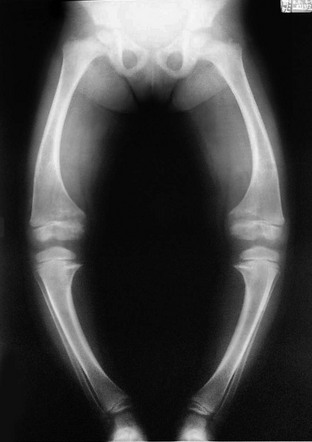
Prominent convex lateral bowing of the femurs and tibias is present, along with rachitic findings in the metaphyses.
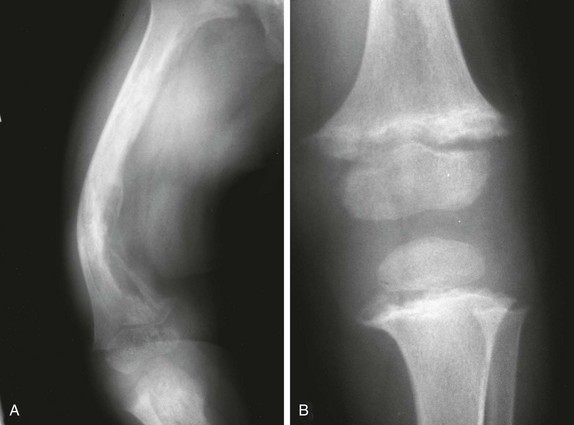
The right femur (A) shows extensive osteolysis. The left hemiskeleton was unaffected by osteolysis, but the left knee (B) shows typical features of rickets.