Chapter 17 Molecular, cellular and tissue effects of radiotherapy
Ionizing radiation, free radical generation, subcellular radiogenic damage
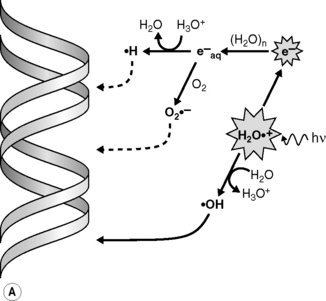
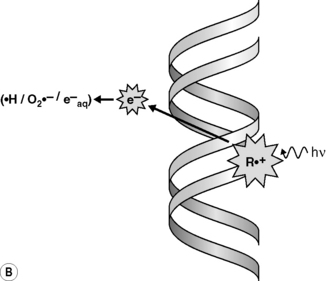
Radiations interact with matter by transferring energy to the molecules of the absorbing material. Indeed, radiations of wavelengths <10−6 cm have sufficient photon energy to eject orbital electrons from the atoms of the absorber’s molecules, leading to their ionization. As the most abundant molecule in a cell is water, the main product of this initial process in a cell is an ionized water molecule (H2O+•); these radical cation species then interact with other water molecules to form hydroxyl radicals (•OH) (Figure 17.1A). The initially ejected electrons (e−) may possess sufficient energy themselves to cause further ionizations until their energy is dissipated and they become solvated to give an aqueous electron (e−aq) and/or combine with other species to generate reducing species such as hydrogen atoms (H•) or superoxide (O2•−). Ionizing radiation is non-discriminatory in that all molecular species in a cell may be damaged. However, DNA is considered to be a key molecular target for the deleterious effects of ionizing radiation in cells. •OH radicals are highly reactive and, together with the less reactive reducing species, may damage DNA via the so-called indirect effect (Figure 17.1A). Radiation can also directly ionize the DNA leading to direct damage of the DNA’s bases or sugar-phosphate backbone (R+•) (Figure 17.1B); although the distinction between direct and indirect damage is not always clear as electrons and radical cations produced in the DNA and DNA-associated water may lead to further ionizations/oxidations in the DNA macromolecule and its surrounding microenvironment.
Through direct and indirect effects, radiation causes a wide range of damage in DNA, including strand breaks, base or sugar damage and cross-links between macromolecules (i.e. DNA–DNA or DNA–protein cross-links) (Figure 17.2A); the frequency of these DNA lesions is shown in Table 17.1. In general, it is considered that the DNA double-strand break (DSB) is the most critical for the lethal effects of radiation; evidence for this is a follows:
• under a variety of experimental conditions, it is the relative level of induced or unrepaired DSBs that best correlates with cell killing
• a single DSB is lethal to yeast
• enzymatically produced DSBs (produced by inserting DNA restriction enzymes into cells) gives the same pattern of chromosome damage and lethality as radiation
• microbeam irradiation has shown the cell nucleus to be the most radiation sensitive site in the cell
• the extreme radiosensitivity of some mutant cell lines is due to defects in DSB repair.
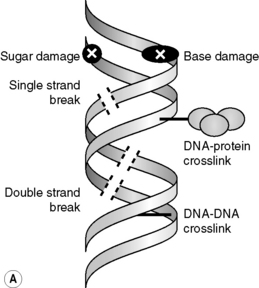
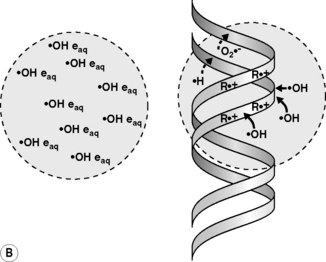
Table 17.1 Types and frequency of radiation-induced damage
| Type of damage | Approximate number per Gy per cell |
|---|---|
| DNA double-strand breaks | 40 |
| DNA single-strand breaks | 1000 |
| DNA–protein cross-links | 150 |
| DNA–DNA cross-links | 30 |
| Base damage | 2000 |
| Sugar damage | 1500 |
However, not all DSBs are the same, as the chemical nature of the strand break ends and the separation between the constituent single strand breaks can vary considerably, as can the proximity of other types of lesions. Indeed, the influence of the proximity of induced lesions has been shown to affect the reparability of damage and this is relevant because low LET radiation (x-rays and γ-rays) can produce multiple ionizations in isolated events (termed spurs and blobs). When these isolated events occur close to or overlap the DNA molecule (see Figure 17.2B), several lesions may be formed within a region of a few base-pairs producing so-called multiply damaged sites (MDS), as described by Ward.
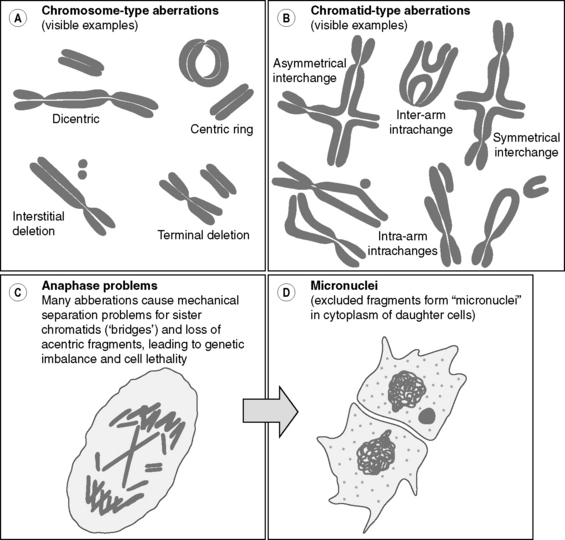
On a larger scale, radiation damage to chromosomes and chromatids leading to aberrations involving breakage and rejoining of chromosome/chromatid fragments (e.g. translocations and ring formations) are observed in many irradiated cells (Figure 17.3). As these events appear to correlate radiation-induced cell killing, such damage is considered an important aspect of the radiation-induced effects in many cells. Furthermore, chromosome damage may be a very sensitive indicator of environmental radiation exposure in an individual.
Recovery, DNA damage repair and cell signalling
DSB repair and cell signalling
Each of the types of radiation-induced DNA damage is repaired by one of several DNA repair pathways (Table 17.2). Of particular significance to the deleterious biological effects of radiation is the repair of DNA DSBs. Consequently, intricate damage response pathways have evolved to repair DSBs. Two principal recombinational DSB repair pathways, that employ mostly separate protein complexes, have been recognized; homologous recombination repair (HRR) and non-homologous end-joining (NHEJ) (Figure 17.4A). Briefly, DSB repair by HRR requires an undamaged template molecule that contains a homologous DNA sequence, typically on the sister chromatid in the S and G2 phases of the cell cycle (see later). In contrast, NHEJ of two double-stranded DNA ends, which may occur in all phases of the cell cycle, does not require an undamaged partner and does not rely on extensive homologies between the recombining ends. It is likely that the balance between NHEJ and HRR in the removal of DSBs depends on the type and location of the lesion. Of note, however, is that NHEJ, unlike HRR, is inherently error prone and possibly mutagenic; that is, this process is potentially unable faithfully to restore the original DNA sequence. Still, NHEJ is considered to be the dominant DSB repair pathway in mammalian cells. The study of these pathways has proved to be a rapidly evolving field of research over the past few years and considerable interest has been generated by the realization that defects in HRR and, in some cases, NHEJ can be causally linked to impaired DNA replication, genomic instability, human chromosomal instability syndromes, cancer development, and cellular hypersensitivity to DNA-damaging agents.
Table 17.2 Pathways involved in the repair of DNA damage
| Repair pathway | Type of lesion | Involved in radiosensitivity? |
|---|---|---|
| Base excision repair (BER) | BD, AP site, SSB | Yes |
| Homologous recombination repair (HRR) | DSB | Yes |
| Non-homologous end joining (NHEJ) | DSB | Yes |
| Nucleotide excision repair (NER) | CL, dimers, bulky adducts | ? |
| Transcription coupled repair (TCR) | BD, dimers, bulky adducts | ? |
| Mismatch repair (MMR) | Mismatched bases | ? |
AP, abasic site; BD, base damage; CL, cross-links; DSB, double strand break; SSB, single strand break.
(From Steel 2002, with permission of Hodder Education.)
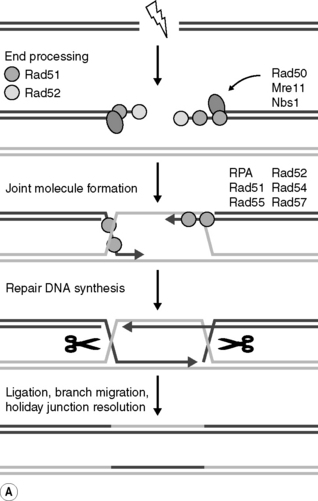
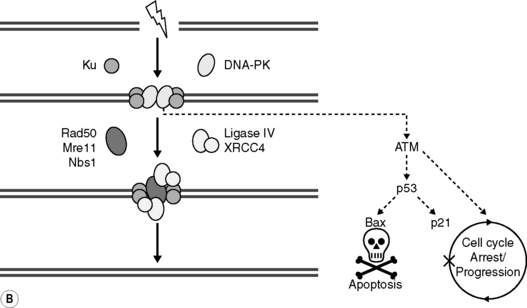
Both NHEJ and HR DSB repair are facilitated by the sequential recruitment of a variety of DNA repair proteins and co-factors to the sites of DNA damage. For example, in NHEJ (see Figure 17.4B), Ku autoantigen, a DNA end-binding heterocomplex of two proteins (Ku70 and Ku80) binds to DSB sends, and then recruits and activates the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to form the active kinase DNA-PK, and possibly the MRE11/RAD50/NBS1 (MRN) complex, whose exonuclease activities may be required for trimming back DNA strands to areas of micro-homology. A DNA polymerase then fills any gaps and the XRCC4/ligase IV heterodimer seals the breaks by forming a phosphodiester bond. Importantly, however, DNA-PK also appears to trigger signal transduction pathways that are involved in stress responses leading to apoptosis (leading to the elimination cells bearing excessive or irreparable damage), cell cycle arrest (allowing cells more time for repair) or cell progression. This is an example of an important concept, developed from studies in molecular radiobiology, that cells possess sensors of induced damage and that these act to link damage to a cell’s ultimate response through intricate cell signaling pathways. Patterns are now emerging that allow critical molecules and pathways to be recognized linking DNA damage and DNA damage repair to classical outcomes of radiation exposure.