Spinal dysraphism
Myelomeningocele/myeloschisis
Lipomyelomeningocele
Meningocele
Split cord malformation or diastematomyelia
Terminal myelocystocele
Caudal regression syndrome
Segmental spinal dysgenesis
Congenital spinal neoplasms such as teratoma, dermoid/epidermoid, hamartomas
Complex syndromes/constellation of anomalies which involve the fetal spine such as OEIS, VACTERL, etc.
Earlier studies of the fetal central nervous system have shown that fetal MRI resulted in a change in diagnosis and management in nearly half of the cases [2, 6]. Multiple studies have been performed to assess utility or fetal MRI in the evaluation of sonographically suspected brain abnormalities [2, 6, 8]. Studies performed have shown that fetal MRI identified additional spinal cord anomalies in 10 % of cases referred for evaluation of sonographically detected bony spinal anomalies [6]. Fetal MRI showed sonographically occult diastematomyelia and segmental spinal dysgenesis [6].
8.4 Fetal MRI in Spinal Dysraphism
Spinal dysraphism is the result of a congenital vertebral defect due to the failure of the caudal neuropore to close which leads to the exposure of the contents of the medullary canal to the exterior. The defect usually involves the posterior vertebral elements, although it can also involve the vertebral body itself. The defect is usually located in the lumbosacral region, but it can occur at any level and it can even involve the entire spine. The absence of skin and muscle in these areas is due to the failure to induct the overlying ectodermal and mesodermal tissues [15].
The increasing use of antenatal US ultrasonography and amniocentesis has resulted in early detection of neural tube defects (NTDs), and the use of fetal MRI has improved accuracy of the diagnosis. Diagnosis of neural tube defects is now evident as early as 18 weeks of gestation, allowing for a time window to have a thorough discussion of the likely outcome with parents.
Spinal dysraphism comprises a wide spectrum of anomalies and can be divided into open and closed defects.
8.5 Open Spinal Dysraphism
Open defects include rachischisis and myelomeningocele. Rachischisis is the most severe form of spina bifida; in this lethal condition, an extensive segment of the rachis is absent, resulting in extensive exposure of neural tissue [16].
8.5.1 Myelomeningocele and Myeloschisis
Myelomeningocele and myeloschisis (myelocele) are the most common open spinal dysraphisms and accounting for 85 % of all cases of spinal dysraphisms [16]. Abnormalities in neurulation can result from defects in disjunction, which involves the process by which there is separation of the neural tube from the overlying ectoderm. Complete failure of a large site of disjunction may result in a myelomeningocele [15].
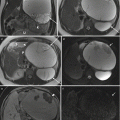
Differentiation of myeloschisis from MMC can be detected on fetal US and fetal MRI. The distinction is based on the position of the neural placode with respect to the level of the skin surface. When there is elevation of the neural placode due to expansion of the subarachnoid space, the lesion is a MMC. In MMC the intramedullary structures lined with meninges protrude or herniate beyond the level of the skin surface (Fig. 8.1). When there is no elevation or protrusion of the neural placode beyond the plane of the skin but a posterior osseous and cutaneous defect is present, it is referred to as a myeloschisis or myelocele. In myeloschisis the spinal canal is directly exposed to the exterior (Fig. 8.2). Besides the aforementioned difference, the imaging findings are identical in MMC and myeloschisis. MMC and myeloschisis are nearly always found with Chiari type II malformation. This association is thought to be due to the collapse of the primitive fetal ventricular system (brought about by the loss of cerebrospinal fluid through the spinal defect), which prevents the rhombencephalic vesicle (from which the brainstem, fourth ventricle, and cerebellum are derived) from expanding and results in deficient development of the posterior fossa and herniation [17].
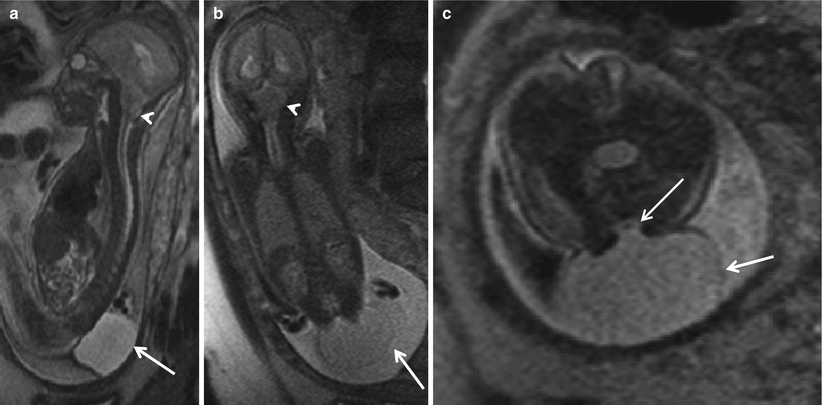
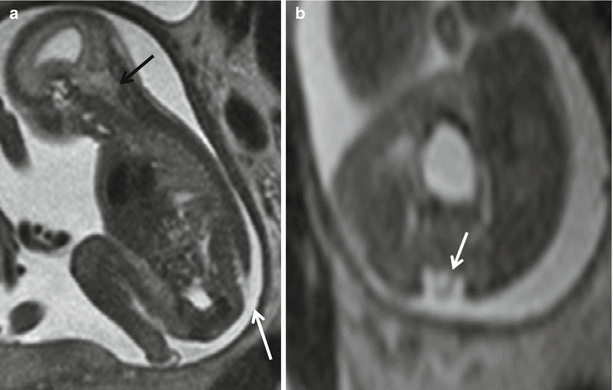
Fig. 8.1
Sagittal (a) and coronal (b) T2 HASTE demonstrating Chiari II with hindbrain herniation (white arrow head) and large thin-walled myelomeningocele sac (white arrow). Axial T2 HASTE (c) – note the low neural placode within the myelomeningocele sac (white arrow)
Fig. 8.2
Sagittal (a) and axial (b) T2 HASTE demonstrating Chiari II with hindbrain herniation (black arrow) and myeloschisis (white arrow). There is no sac with the neural placode (white arrow) exposed to the amniotic fluid
The stigmata of Chiari II malformation include a small posterior fossa, beaking of the tectum, hindbrain herniation, and effacement of the supratentorial CSF spaces. Additional findings of dysgenetic corpus callosum, and subependymal gray matter heterotopia may be present. Ventriculomegaly may be seen in many of these cases. Both US and MR can visualize the direct and indirect stigmata of Chiari II malformation such as fetal cranium (the lemon sign: special shape of the cranium due to deformity of the frontal bones) and the banana sign (special shape of the posterior fossa secondary to displacement of the cerebellar hemispheres toward the cervical canal), which results in the obliteration of cerebrospinal fluid at the foramen magnum [17]. The size of the defect and of the sac in MMC is variable and could be small or large (Fig. 8.3). MR can demonstrate the neural placode elements entering into the sac. Detection of presence of hindbrain herniation is ready apparent with fetal MRI but may be sometimes difficult to detect with screening US. Excellent detail and soft tissue resolution can be obtained with MRI, but fetal US may be more accurate in determining the level of the defect and determination of the neural placode, making the two modalities complementary [1, 18].
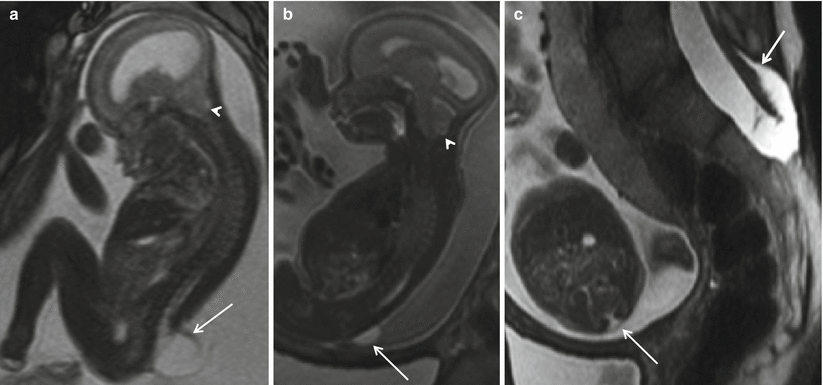
Fig. 8.3
Sagittal (a) T2 HASTE demonstrating Chiari II with hindbrain herniation (white arrow head) and large myelomeningocele sac (white arrow). Sagittal (b) and axial (c) T2 HASTE images from another patient demonstrating small myelomeningocele sac (white arrow). Mother also had myelomeningocele repaired postnatally (white arrow)
An extremely rare type of open dysraphism is the hemimyelomeningocele or hemimyelocele: in this anomaly, diastematomyelia is associated with one of the abovementioned anomalies in which one of [15] the hemi cords does not complete neurulation [15]. This entity has been discussed separately in the later part of this chapter.
In addition to the standard options of termination of the pregnancy and continuation of the pregnancy until term with cesarean or vaginal delivery, fetal closure of the dysraphic defect as part of the management of myelomeningocele study (MOMS) is now an option for some families in the United States [9, 10].
The results of the MOMS trial convincingly demonstrated that the cases treated with prenatal MMC closure had a decreased incidence of hindbrain herniation [10, 19]. The ascent of hindbrain structures could be demonstrated within 3 weeks of the fetal closure using serial MRI (Fig. 8.4). The overall fetal head size and posterior fossa volume have been demonstrated to be small in MMC patients and have been shown to increase normally after fetal surgery [10] (Fig. 8.5). The return of the head size to normal is likely attributed to restoration of cerebrospinal fluid volume after reversal of hindbrain herniation [10]. Recently published results from the MOMS study indicate that prenatal fetal MMC repair leads to reversal of hindbrain herniation, improvement in the lower extremity function, and decreased need for CSF diversion compared with postnatal repair controls [10, 19, 20].
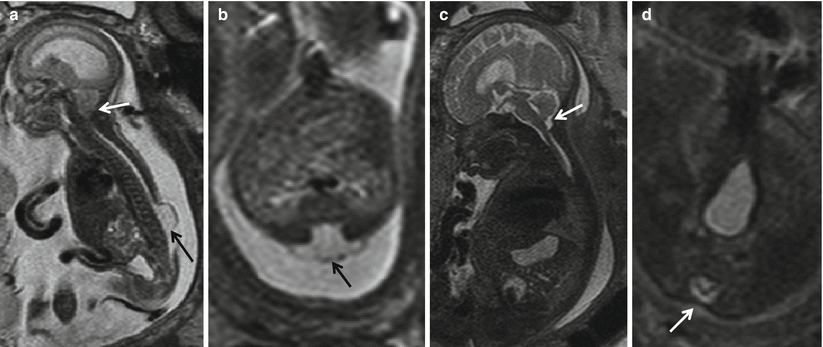
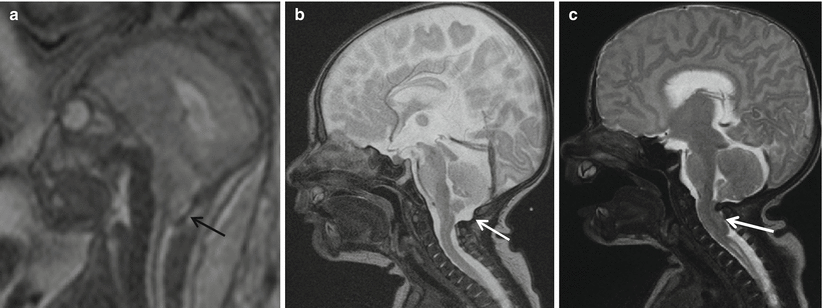
Fig. 8.4
(a, b) Pre-repair fetal MRI at 24 weeks and (c, d) post-repair fetal MRI at 30 weeks show that the sac (black arrow) has been resected and the defect closed (white arrow). Note the resolution of hindbrain herniation and CSF seen at the CV junction on the post-repair fetal MRI (white arrow)
Fig. 8.5
(a) Pre-repair fetal MRI and (b) post-fetal-repair postnatal MRI show the resolution of hind brain herniation and CSF seen at the CV junction (white arrow). In comparison with postnatal repair of MMC (c), there is persistent hindbrain herniation and effaced CSF at the CV junction (white arrow)
In our institute, the fetus was only considered for prenatal myelomeningocele surgery if the gestational age at the time of the proposed surgery was 26 weeks or less, if the transatrial ventricular dilation was less than 16 mm (normal being less than 10 mm), if the estimated level of the lesion was at or above S1, and if there was convincing leg and foot motion on ultrasound in the absence of foot or leg deformity. To provide all this accurate information, fetal MRI was an integral part of the MOMS trial workup prior to prenatal surgery [10]. The discussion of the MOMs trial and fetal myelomeningocele repair in detail is beyond the scope of this chapter. Excellent papers on this topic are available to the interested reader [10, 18–25].
8.6 Closed Spinal Dysraphism
Closed dysraphism accounts for about 15 % of cases of spinal dysraphism [15]. The closed dysraphic defects tend to be small lesions and they are completely covered with skin. When there is a premature disjunction of the cutaneous ectoderm from the neuroectoderm, it allows mesenchyme to contact the inner portion of the developing neural tube. As the neural tube begins to close, the mesenchymal gets inducted to become fat, which can interfere with neurulation. This results in the terminal lipoma or lipomyelomeningocele/lipomyeloschisis. Typically these skin-covered lesions are not associated with Chiari II malformation or ventriculomegaly and do not manifest with elevated alpha-fetoprotein [26].
Closed spinal dysraphism can be subdivided into lesions with subcutaneous masses and those without. There are three main types of closed spinal dysraphisms with masses. The first type is further subdivided into lipomyelomeningocele and lipomyeloschisis (lipomyelocele). The placode-lipoma interface with respect to the plane of the back determines the distinction between lipomyelomeningocele and lipomyeloschisis. The interface in a lipomyelomeningocele is outside the neural canal due to the expansion of the subarachnoid space, whereas in lipomyeloschisis, the lipoma joins the neural placode inside the neural canal after entering through a defect in the bone (Figs. 8.6 and 8.7). The placode in the lipomyelomeningocele is likely to be deformed and rotated towards the lipoma and away from the protrusion of the meninges [15, 16].
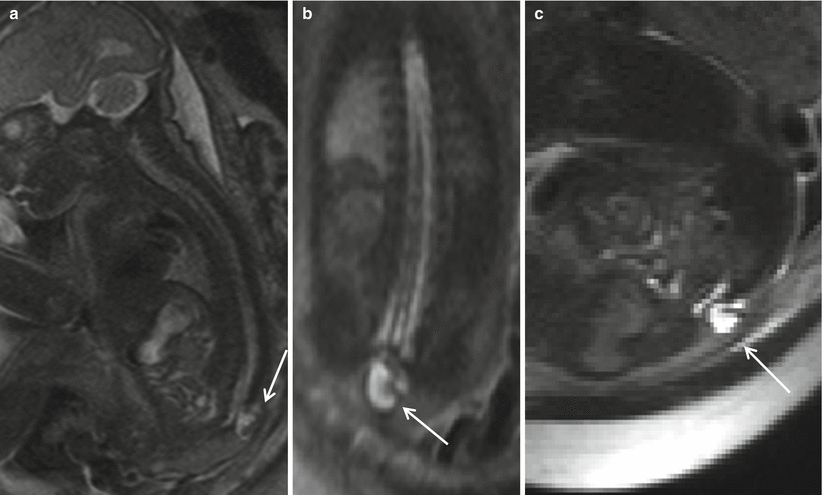
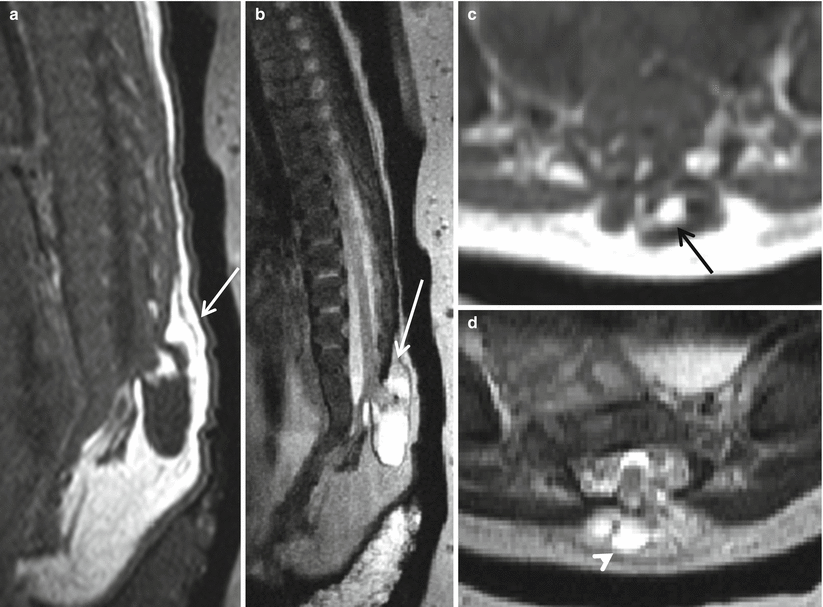
Fig. 8.6
Sagittal (a), coronal (b), and axial (c) T2 HASTE demonstrating skin-covered sac, protruding through posterior defect beyond the spinal canal in the lumbosacral region, containing neural elements suggesting a lipomyelomeningocele sac (arrow). Note the lack of hindbrain herniation.
Fig. 8.7
Sagittal T1 (a), sagittal T2 (b), axial T1 (c), and axial T2 (d) postnatal imaging features of lipomyelomeningocele with a skin-covered dysraphic defect (white arrow), anteroposteriorly split neural placode of the distal low-lying spinal cord, with presence of fat (black arrow) and CSF (white arrowhead). Hypoplastic sacrum and coccyx
The second type of closed spine dysraphism with masses is a meningocele. These lesions are herniated sacs of cerebrospinal fluid lined with meninges and skin and traditionally thought not to contain neural elements. Meningoceles are usually posterior herniations but may be lateral or anterior. Meningoceles are most commonly located in the thoracic and lumbar spine (Fig. 8.8). Anterior meningoceles are much less common and tend to be located in the presacral region [27]. The association of bony anomalies is rare and often mild. The role of prenatal imaging is to distinguish meningocele from myelomeningocele.
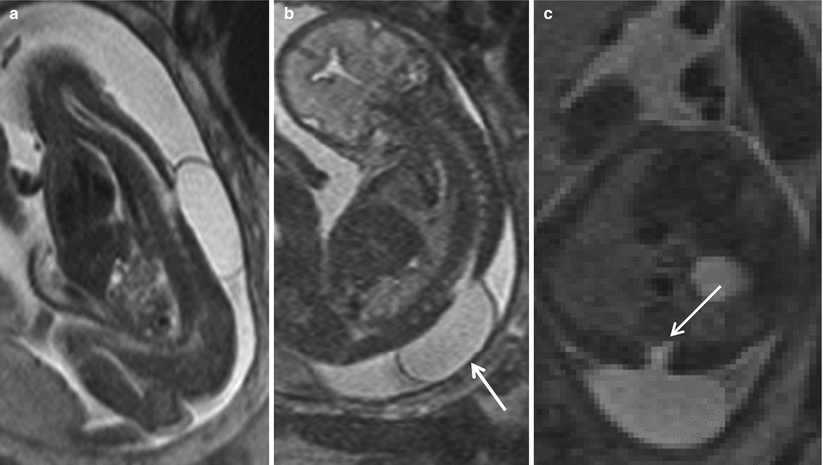
Fig. 8.8
Sagittal (a), coronal (b), and axial (c) T2 HASTE show a CSF-containing sac protruding from a defect in the lower thoracic spine (white arrow) consistent with a meningocele. Neural elements (white arrow) do not extend into the meningocele
The third type of closed spine dysraphism with masses is a myelocystocele. These lesions can occur at any point in the spine. When the myelocystocele involves the most distal spine, it is referred to as a terminal myelocystocele. Myelocystoceles result from marked dilatation of the expanded central canal of the spinal cord which herniates through the lumbosacral dysraphic defect (Fig. 8.9). The leptomeninges herniate around the distended spinal cord. Some consider a terminal myelocystocele to represent a severe form of the dilatation of persistent terminal ventricle due to trapping of the CSF within the neural tube during formation. This entity is characterized by thick wall of the protruding sac. Myelocystoceles can be differentiated from a myelomeningocele by the thickness of the wall of the herniated sac, the absence of the Chiari II malformation, and the absence of elevated alpha-fetoprotein levels [26, 28]. Myelocystoceles can be associated with complex cloacal anomalies [15, 29] (Fig. 8.10).
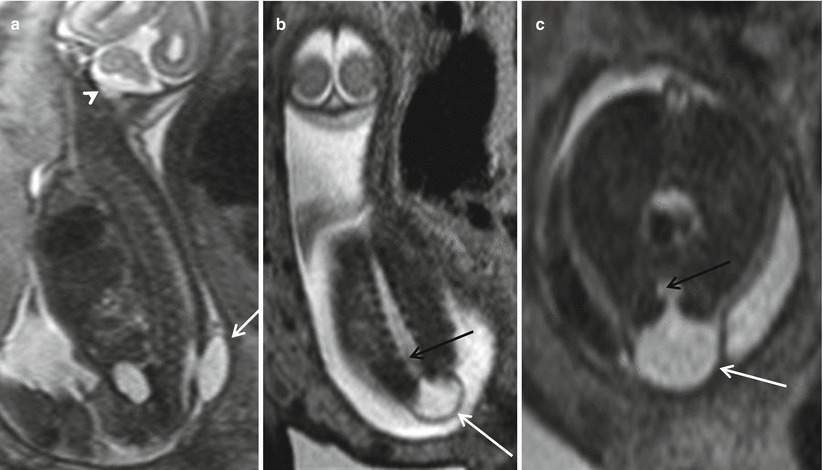
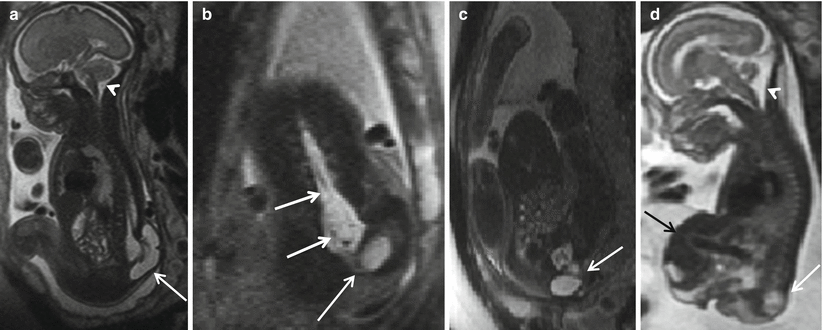
Fig. 8.9
Sagittal (a), coronal (b), and axial (c) T2 HASTE demonstrate a thick-walled skin-covered sac with CSF content (white arrows) herniating from a defect in the lower lumbar spine. There is continuation of the central portion of the distal cord into the sac (black arrow). Note the lack of hindbrain herniation (white arrowhead)
Fig. 8.10
Sagittal (a, d), coronal (b), and axial (c) T2 HASTE reveal spinal dysraphism involving almost entire length of the lumbar spine down to sacrum with a large terminal myelocystocele (white arrows). No hindbrain herniation (white arrowhead). Note continuation of the dilated central canal of the low spinal cord into the cyst consistent with a terminal myelocystocele (white arrows). There is a giant fetal omphalocele containing a majority of the liver and gallbladder (black arrow). Possible bladder exstrophy and imperforate anus
A thick-walled sac, absence of the hindbrain herniation, and lack of the elevation of maternal or amniotic fluid alpha-fetoprotein should raise suspicion of an occult dysraphism such as a meningocele, lipomyelomeningocele, or myelocystocele [3].
Closed spinal dysraphisms without subcutaneous masses can be divided into simple and complex anomalies. Simple lesions include intradural lipomas, filum terminale lipomas, tight filum terminale syndrome, persistent terminal ventricle, and dermal sinus. Detecting all these findings prenatally is extremely difficult.
When the failure of disjunction is at very small site, it results in a dermal sinus. Prenatal diagnosis of an isolated small dermal sinus is extremely difficult. However, when there is a large track which runs from the skin surface through the subcutaneous fat, it may be evident with US and MR imaging.
Complex closed spinal dysraphisms can be divided into disorders of midline notochord integration and disorders of notochord malformation. Disorders of midline notochord integration include enteric fistula, which is an abnormal connection between the digestive tract and the surface of the skin that may or may not be accompanied by a neurenteric cyst. Neurenteric cysts are lined with epithelial mucosa similar to that of the gastrointestinal tract and are typically located in the anterior cervicothoracic region.
Another disorder of midline notochord integration includes split cord malformation, or diastematomyelia (Fig. 8.11). The cord is focally divided into two hemi cords, often separated by a bony bar or a fibrous septum. This splitting may involve the entire anteroposterior aspect of the cord or only a portion of the spinal cord. Each hemi cord contains a central canal, one dorsal horn, and one ventral horn, from each of which there is origin of one nerve root. Division between the two dural tubes is by a bony bar (type I) or division by a fibrous septum (type II). Sometimes there may be no intervening septum in the type II lesion [30]. Diastematomyelia most commonly occurs in the lumbar region and is associated frequently with vertebral body anomalies [15, 30]. Type II diastematomyelia is easily recognized fetal MR imaging. EPI techniques are useful in identifying the separating bony bar. It has been reported that diastematomyelia may be present in up to 40 % of MMC cases [3].
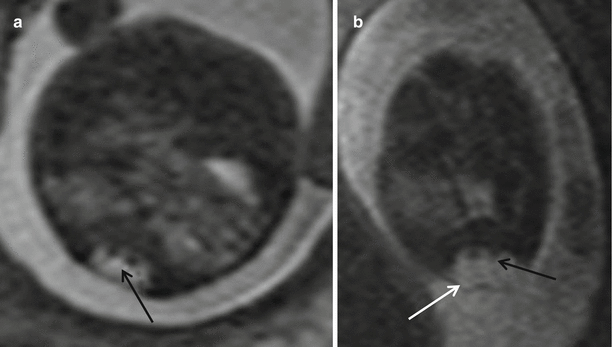
Fig. 8.11
(a) Axial T2 HASTE showing split cord malformation – diastematomyelia (black arrow). (b) shows diastematomyelia within a myelomeningocele (white arrow)
It is important for the interpreting radiologist to perform a diligent search for the presence of diastematomyelia in every case of MMC. This finding is important because if diastematomyelia is not suspected, the surgical repair of the myelomeningocele will not include the repair of the tethered cord that is nearly always present in diastematomyelia [9, 31].
8.7 Segmental Spinal Dysgenesis (SSD)
Another entity of congenital spinal abnormalities includes the segmental spinal dysgenesis (SSD). This is a rare abnormality in which a focal segment of the lumbar or thoracic spine is absent or markedly hypoplastic, and the spinal cord is segmentally disrupted. The distal spinal cord is abnormally large although the distal spinal canal is unaffected. Above the affected level, the spine, spinal canal, and spinal cord are normal. It has been hypothesized that this discrete demarcation suggests a segmental malformation or a focal insult to a previously normal spine. SSD is characterized by associated marked kyphosis and anomalous lower extremities. On MR imaging, SSD findings include marked hypoplasia of one or more vertebral segments and associated hypoplasia of the corresponding segments of thecal sac and spinal cord [32].
Some authors consider SSD as to be a part of the spectrum of caudal regression syndrome with the notochordal lesion occurring more proximally. If the notochordal development is affected distally, then caudal regression syndrome is the result. The incidence of caudal regression syndrome is higher than SSD (reported to be 11:1 according to one series) [32]. This high incidence of caudal regression syndrome suggests that there is a higher degree of susceptibility of the caudal cell mass for developmental derangement.
8.8 Caudal Regression Syndrome
Caudal regression syndrome is a disorder of notochord malformation, which consists of the total or partial agenesis of the lumbar and sacral spine, probably secondary to a disturbance in the formation of the secondary neural tube [33].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


