Chapter 80 Cardiomyopathy is a chronic and often progressive disease of the myocardium with associated cardiac dysfunction. It is a rare but serious disorder; only 25% of children survive more than 5 years after the onset of symptoms. Although our understanding of the causes of pediatric cardiomyopathy has advanced, the prognosis has not changed considerably in the past 30 years and is the same in developing and industrialized nations.1,2 Cardiomyopathy can be classified according to the dominant pathophysiology and etiology (e-Box 80-1). Four major physiologic forms of cardiomyopathy exist: dilated, hypertrophic, restrictive, and arrhythmogenic. Hypertrophic and dilated cardiomyopathies are the most common forms in children.2 Patients may be classified as having more than one physiologic form of cardiomyopathy. Cardiomyopathy also may be classified according to etiology. Cardiomyopathies associated with particular cardiac or systemic diseases are referred to as specific cardiomyopathies. Inflammatory and isolated familial cardiomyopathies account for the majority of pediatric cases with a known cause.3 Other specific cardiomyopathies that are less common in the pediatric population include those associated with metabolic disorders, general systemic diseases, muscular dystrophies, neuromuscular disorders, and sensitivity or toxic reactions (e.g., radiation and chemotherapy). In two thirds of children with cardiomyopathy, no etiology is found.2,3 Because of its availability, lack of radiation use, and portability, echocardiography is the most common method used to classify cardiomyopathy and evaluate cardiac function. With improvements in technology, cardiovascular magnetic resonance imaging (MRI) has become an accepted tool for assessing cardiomyopathy.4,5 With the multiple techniques available, cardiac MRI can be used to assess myocardial morphology and function (cine sequences), myocardial perfusion reserve (first-pass contrast-enhanced perfusion), and myocardial viability and scar formation (delayed enhancement sequence).4 Hypertrophic cardiomyopathy is commonly familial and is inherited in an autosomal-dominant fashion with variable penetrance and expression.6 Although some children are asymptomatic, hypertrophic cardiomyopathy can present with arrhythmias and sudden cardiac death at any age.4 Hypertrophic cardiomyopathy generally is characterized by left ventricular hypertrophy without a demonstrable cause. Although any region of the left ventricle can be involved, hypertrophy of the interventricular septum often is present, which may cause left ventricular outflow tract obstruction (Fig. 80-1 and Video 80-1). The right ventricle also may be affected. Figure 80-1 Hypertrophic cardiomyopathy. Echocardiography is the standard method of diagnosing hypertrophic cardiomyopathy, but cardiac MRI can be used to assess the location and degree of hypertrophy, including areas difficult to visualize with echocardiography, such as the cardiac apex and portions of the right ventricle.7 Cine gradient echo sequences can be used to evaluate the flow dynamics of the left ventricular outflow tract both before and after surgery. Contrast-enhanced MRI also may demonstrate delayed enhancement within scarred or fibrotic areas of myocardial hypertrophy—areas thought to play a role in the arrhythmias associated with hypertrophic cardiomyopathy.8,9 Dilated cardiomyopathy is characterized by dilation and impaired contraction of the left ventricle or both ventricles, and patients typically present with progressive heart failure.6 Most pediatric cases are idiopathic, but causes include infectious myocarditis, familial disease, and neuromuscular disorders (Duchenne and Becker muscular dystrophies).10 Coronary artery disease, a common cause of dilated cardiomyopathy in adults, is uncommon in childhood, although in children with sickle cell disease, ischemic cardiomyopathy related to microinfarctions may develop.1,11 Most children with dilated cardiomyopathy are diagnosed in the first year of life.10 The functional and anatomic changes associated with dilated cardiomyopathy can be well evaluated with cardiac MRI, including acute and chronic changes of myocarditis and changes in left ventricular mass, stroke volume, ejection fraction, and myocardial late enhancement.5 Arrhythmogenic right ventricular cardiomyopathy (ARVC) is characterized by progressive fibrofatty replacement of right ventricular myocardium. This tissue replacement starts focally in the right ventricle but may progress to involve the entire right ventricle; it may involve the left ventricle as well. Areas of spared myocardium may act as foci of instability, resulting in ventricular arrhythmias and sudden death, which become more common in adolescents and young adults.12 The fatty or fibrofatty myocardial infiltration and wall thinning of ARVC can be visualized on T1-weighted spin echo magnetic resonance (MR) sequences (Fig. 80-2). The most reliable MR findings include abnormal right ventricular regional and global wall motion, aneurysms, and dilation. Late gadolinium enhancement can also be seen.5,13,14 Figure 80-2 Arrhythmogenic right ventricular cardiomyopathy in an 18-year-old with recurrent supraventricular tachycardia. Restrictive cardiomyopathy, the least common type, is characterized by restricted filling and decreased diastolic volume of one or both ventricles, without ventricular dilation or hypertrophy.6 Restrictive cardiomyopathy is usually idiopathic and may be associated with diseases that cause infiltration or fibrosis of the myocardium or endocardium, such as hemochromatosis, glycogen storage diseases, and Gaucher, Hurler, and Fabry diseases.12 Although amyloidosis and sarcoidosis are rare in children, they also may cause restrictive cardiomyopathy. Because both ventricles may be involved, patients may present with signs and symptoms of right or left ventricular failure or arrhythmias.4 Restrictive cardiomyopathy should be considered in patients presenting with heart failure but without cardiomegaly or systolic dysfunction. It is important to distinguish restrictive cardiomyopathy from constrictive pericarditis, which has a similar clinical appearance but can be cured surgically.15 Radiographs of the chest in patients with restrictive cardiomyopathy may demonstrate a normal heart size. Pulmonary congestion, interstitial edema, and pleural effusions may be seen.15 Cardiac MRI may demonstrate ventricles that are small to normal in size (Fig. 80-3); signs of poor ventricular filling may be present, including dilation of the atria, superior and inferior venae cavae, and hepatic veins.4 These findings are similar to those of constrictive pericarditis, but the pericardial thickening (>4 mm) typical of constrictive pericarditis is absent.16 Figure 80-3 Restrictive cardiomyopathy. The inflammatory cardiomyopathies include both infectious and familial forms. Among cases with a known cause, nearly 30% are attributed to infection.3 Although infectious cardiomyopathy, also known as myocarditis, can be caused by bacterial, fungal, or parasitic infection, viral myocarditis is most common. Enteroviruses, particularly Coxsackie virus B, are associated with 25% to 40% of cases of pediatric acute myocarditis and dilated cardiomyopathy.17 The acute clinical presentation of myocarditis is characterized by dyspnea and tachypnea due to congestive heart failure. Myocardial involvement by tuberculosis is extremely rare and typically occurs in the setting of disseminated disease.18 Eosinophilic myocarditis may be seen in the setting of eosinophilic syndromes or allergic reactions.19 Acute rheumatic fever, discussed later in more detail, is an autoimmune response that follows a small percentage of infections with group A β-hemolytic streptococcus. The sequela, rheumatic heart disease, is significant because of the associated morbidity that may include valvular disease (more common) and carditis (pericarditis, myocarditis, or endocarditis) in the acute setting.20 Isolated familial cardiomyopathy typically is defined as cardiomyopathy with no systemic features occurring in a patient with an identified genetic defect or in multiple family members. Familial cardiomyopathy accounts for approximately 20% to 25% of nonidiopathic cases of cardiomyopathy.3,21 Sarcoidosis is a multiorgan disease characterized by noncaseating granulomas. It occurs most often in adolescents but has been reported in children as young as 2 years. Clinical manifestations vary according to age. In children younger than 5 years, the disease involves mainly the skin, eyes, and joints, whereas in older children, involvement of the lymph nodes, lungs, or eyes is more common.22 Cardiac disease is uncommon but has a wide spectrum of manifestations, including conduction abnormalities (usually ventricular arrhythmias) and heart block. Infiltration of the ventricular walls can compromise ventricular contraction and compliance, resulting in systolic and diastolic failure, respectively. Other cardiovascular manifestations include pericardial effusion, papillary muscle dysfunction, and valvular disease.22 MRI has been used to make the diagnosis of cardiac involvement with infiltration of the myocardium. Neuromuscular cardiomyopathies include a variety of genetic and acquired disorders. Duchenne muscular dystrophy, the most common of the muscular dystrophies, is an X-linked recessive neurodegenerative disorder characterized by progressive skeletal muscle weakness. In children with Duchenne muscular dystrophy, dilated cardiomyopathy develops with alternating areas of hypertrophy, atrophy, and myocardial fibrosis.23 Interestingly, dilated cardiomyopathy may be the only clinical manifestation of genetic carriers of Duchenne muscular dystrophy.24 Friedreich ataxia is a rare, autosomal recessive neurologic disorder characterized by progressive ataxia and musculoskeletal abnormalities (scoliosis and foot deformities). The cardiac disease of Friedreich ataxia includes hypertrophic cardiomyopathy that progresses to dilated cardiomyopathy, along with arrhythmias. Although patients typically present with neurologic symptoms, a small minority may present with symptoms of systolic dysfunction.25 Noonan syndrome is a relatively common genetic syndrome characterized by typical facies and short stature. Cardiac abnormalities associated with Noonan syndrome include pulmonary valve stenosis and hypertrophic cardiomyopathy, with or without obstruction.26 The glycogen storage diseases (GSDs) constitute a group of rare diseases characterized by abnormal glycogen breakdown, with resultant storage in various tissues. Cardiac involvement is most common in types II (Pompe disease), III, and IV.27 Pompe disease (GSD type II), which is associated with a deficiency of acid α-glucosidase, has three major forms. A complete deficiency of acid α-glucosidase leads to the most severe infantile form, with progressive lethal cardiac and muscle disorder. Symptoms include failure to thrive, hypotonia, respiratory difficulties, and cardiac problems. The cardiac features include congestive heart failure, arrhythmias, cardiomegaly, biventricular hypertrophy, and outflow tract obstruction.28 Marked cardiomegaly, with or without congestion, in an infant beyond the immediate newborn period should raise the possibility of this entity as a differential diagnosis (Fig. 80-4).27 Partial deficiencies of acid α-glucosidase lead to milder, late-onset juvenile and adult forms of Pompe disease. Figure 80-4 Glycogen storage disease of the heart (Pompe disease) in a 5-month-old girl. Nearly all patients with GSD type III have clinically silent heart disease. Patients with GSD type IV typically die of liver disease before their cardiomyopathy becomes clinically obvious. Echocardiography demonstrates cardiac hypertrophy, which may result in left ventricular outflow tract obstruction, but no cardiac dilation.27
Myocardial, Endocardial, and Pericardial Diseases
Myocardial Diseases
Hypertrophic Cardiomyopathy
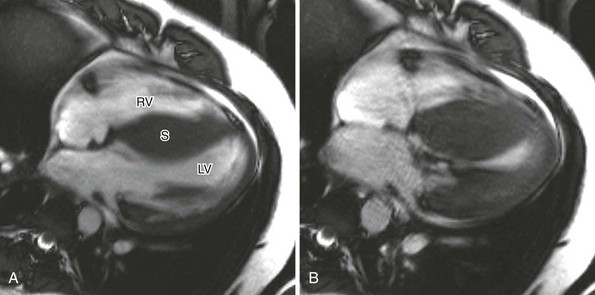
A, Four-chamber cine magnetic resonance view at end-diastole shows marked septal (S) thickening. B, During systole, near-complete obliteration of the left ventricular chamber occurs. LV, Left ventricle; RV, right ventricle. (Courtesy Laura Heyneman, Duke Medical Center.)
Dilated Cardiomyopathy
Arrhythmogenic Right Ventricular Cardiomyopathy
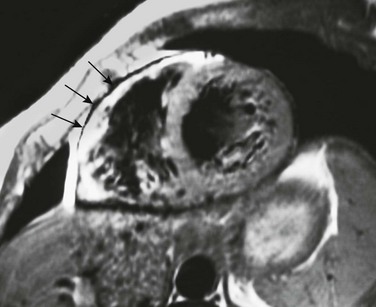
A T1-weighted short-axis image through the heart shows an abnormal high signal involving the anterior wall of the right ventricle (arrows) adjacent to the normally high T1 signal of pericardial fat. This appearance is consistent with the fibrofatty myocardial replacement and wall thinning seen with arrhythmogenic right ventricular cardiomyopathy.
Restrictive Cardiomyopathy
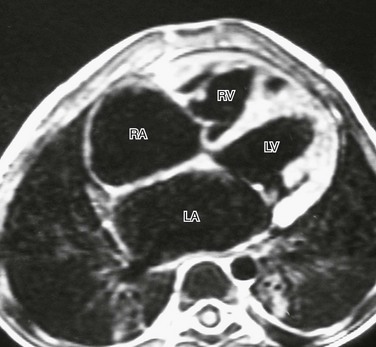
A T1-weighted four-chamber view in a 3-year-old boy with idiopathic restrictive cardiomyopathy demonstrates the relatively small size of the right (RV) and left (LV) ventricles, with dilation of the right atrium (RA) and left atrium (LA). There is no evidence of pericardial thickening to suggest restrictive pericarditis.
Specific Cardiomyopathies
Inflammatory Cardiomyopathy
Sarcoidosis
Neuromuscular Cardiomyopathy
Metabolic Cardiomyopathy
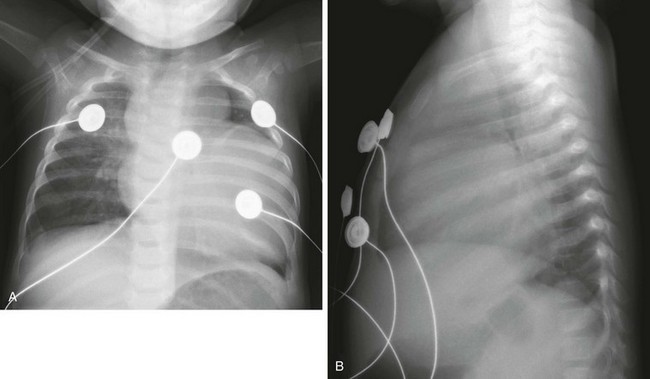
Marked cardiomegaly is seen on frontal (A) and lateral (B) radiographs. The prominent lobular shape of the superior mediastinum is due to the thymus.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Myocardial, Endocardial, and Pericardial Diseases