Chapter 38 Epilepsy is a common pediatric neurological disorder. In North America, the overall annual incidence of epilepsy is approximately 50 per 100,000 persons. The incidence is highest for children younger than 5 years and for elderly persons.1,2 Children are at higher risk for the development of epilepsy than are adults. Atypical, idiopathic, and focal epilepsy, as well as epileptic syndromes, require evaluation with magnetic resonance imaging (MRI). In approximately 30% of children with epilepsy, the disease becomes refractory to medical therapy.3 In children with refractory epilepsy, neuroimaging is critical for identification of an epileptogenic substrate that is responsible for the epilepsy, particularly in children undergoing surgery. Patients without a lesion (that is, patients who have normal MRI findings) have been reported to have poorer outcomes of epilepsy surgery compared with patients who have a lesion identified upon neuroimaging.4 Epileptogenic substrates include malformations of cortical development (MCDs), developmental tumors, anoxic-ischemic injuries, prior cerebrovascular disease, neurocutaneous syndromes, and Rasmussen encephalitis. Concurrent lesions such as MCD, hippocampal sclerosis, and developmental tumors can occur in approximately 13% to 20% of cases.5,6 Another important role of imaging in children with intractable epilepsy is to identify the location of eloquent cortex and white matter tracts as part of the presurgical evaluation (see Chapter 27). The MRI epilepsy protocol should include volumetric T1-weighted imaging, T2-weighted imaging, fluid attenuated inversion recovery (FLAIR), proton density, and inversion recovery sequences in at least two orthogonal planes, covering the entire brain. Volumetric gradient echo T1-weighted acquisitions with 1- to 1.5-mm section thickness provide excellent gray/white matter contrast and are useful for better anatomic delineation. This sequence can be reformatted into any orthogonal or nonorthogonal plane without the penalty of increased scan time and can be used for anatomic integration of functional data, stereotactic electrode placement, and neuronavigation.7–9 In persons with temporal lobe epilepsy, the coronal plane should be acquired perpendicular to the long axis of the hippocampus. Injection of contrast material does not improve the detection of a lesion but may help characterize a lesion once it is found. In cases in which an epileptogenic focus is not identified, further evaluation with dedicated higher resolution MRI, image postprocessing, or additional imaging techniques including diffusion tensor imaging, magnetic resonance spectroscopy, and functional imaging with interictal positron emission tomography (PET) or ictal/interictal single photon emission computed tomography (SPECT), along with magnetoencephalography, may help in identifying a lesion or the epileptogenic zone.10,11 MCDs are a major cause of drug-resistant epilepsy,12 in particular focal cortical dysplasia (FCD), hemimegalencephaly, and tuberous sclerosis. Other MCDs such as lissencephaly, gray matter heterotopia, polymicrogyria, and schizencephaly also are associated with epilepsy.13–16 Overview: FCD is an MCD that is intrinsically epileptogenic. It is one of the most common causes of intractable epilepsy in children and accounts for up to 39% of surgical cases.17,18 The mechanism of epilepsy is still unclear. Possibilities include abnormal firing from the dysplastic neurons rather than from balloon cells,19 dysfunction of synaptic circuits with abnormal synchronization of the neuronal population, and abnormal organization of the inhibitory interneurons.20 Numerous classifications of FCD have been proposed.13,21–23 Recently, a consensus classification has been proposed by the International League Against Epilepsy (Table 38-1).24 Table 38-1 Histopathologic Classification of Focal Cortical Dysplasia FCD, Focal cortical dysplasia. From Blumcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52(1):158-174. Imaging: FCD can be located in any cortex of the cerebral hemisphere and can have variable size, from one gyrus to more than one lobe. The MRI features of FCD are increased cortical thickness, blurring of the cortical–white matter junction, increased T2 or FLAIR signal in the cortex and subcortical white matter, high T1 signal in the cortex, and an abnormal sulcation and gyration pattern. Taylor FCD or FCD type IIB more commonly involves the extratemporal cortex and is more likely to demonstrate a high T2 and FLAIR signal that tapers toward the ventricle.21,25,26 Non-Taylor FCD (type I FCD and mild MCDs) is more likely to be located in the temporal lobe and to demonstrate hypoplasia or atrophy of the white matter and mild increased signal. FCD, in particular non-Taylor FCD, is associated with hippocampal sclerosis.27 The MRI appearance of FCD may change with brain maturation. Longitudinal MRI studies in infants with FCD have shown that findings of the early study may be normal but later imaging may demonstrate a high T2 signal in the white matter, a high T1 signal in the cortex, and blurring of the cortical/subcortical white matter junction.28 The high signal in the white matter may be a result of abnormal myelination, either because of the underlying disease or as a result of seizures. Thus even when the MRI findings appear normal in a neonate or an infant with refractory partial seizures or infantile spasm, a repeat study is recommended (Fig. 38-1).28 In contrast to the high T2/FLAIR signal in the white matter of FCD in children, the white matter adjacent to the dysplastic cortex may demonstrate a low T2 and high T1 signal in neonates and infants, which is thought to be secondary to early white matter myelination as a result of repeated seizures.29 Figure 38-1 A 5-year-old girl with focal cortical dysplasia. Treatment: Good seizure control or a seizure-free outcome is achieved in up to 50% to 70% of patients with FCD after surgical resection of the FCD.30–32 This outcome compares favorably with the outcome of patients who have hippocampal sclerosis33 and a low-grade neoplasm.34 The reported surgical outcomes of persons with subtypes of FCD are variable. Authors of some studies reported a better surgical outcome in persons with Taylor FCD,21,35,36 whereas other authors reported better surgical outcomes in persons with other subtypes of FCD, including type I FCD and mild MCD.37,38 Differences in outcomes of persons with subtypes of FCD may in part be related to the preponderance of type I FCD and mild MCD in the temporal lobe.37 Overview: Hemimegalencephaly is an MCD that is characterized by one hemisphere being larger than the contralateral side. It may be sporadic or it may be associated with a variety of syndromes, including proteus syndrome, epidermal nevus syndrome, hypomelanosis of Ito, linear nevus sebaceous syndrome, neurofibromatosis type I, and tuberous sclerosis. The sporadic form is considered a hemispheric variant of FCD.39 On histology, the appearance is similar to FCD with abnormal gyration of the cortex, dyslamination, blurring of the gray/white matter junction, giant neurons in both gray and white matter, and balloon cells in 50% of cases. The most common clinical presentation is early intractable epilepsy; other clinical presentations include hemiparesis, hemianopia, and mental retardation. Imaging: The affected hemisphere is larger than the contralateral side. The cortex is dysplastic and thick, with broad gyri and shallow sulci (e-Fig. 38-2). The ipsilateral lateral ventricle may be enlarged. The ipsilateral white matter demonstrates variable signal changes, depending on the age of the patient. Neonates and infants usually demonstrate a high T1 and low T2 signal in the white matter suggestive of early myelination, whereas in older children the white matter shows a low T1 signal and a high T2 signal with associated cystic changes and calcification.40–42 With recurrent seizures or status epilepticus, the enlarged hemisphere later may become atrophic.43 e-Figure 38-2 A 2-year-old with hemimegalencephaly. Overview and Imaging: Tuberous sclerosis complex is an autosomal-dominant neurocutaneous syndrome characterized by multisystem involvement including the brain, eyes, heart, kidney, skin, and lung. Seizures occur in 80% to 90% of patients, and seizures are intractable in 25% to 30% of patients.44 Children with medically refractory epilepsy usually have multiple cortical/subcortical tubers that exhibit broad gyri, a thick cortex, and an abnormal signal in the cortex and subcortical white matter. The cortical/subcortical tubers occasionally may demonstrate calcification and cystic degeneration. A combination of structural and functional imaging such as fluorodeoxyglucose (FDG)-PET, ictal/interictal SPECT, and magnetoencephalography can be used to identify the epileptogenic tubers45–48 (Fig. 38-3). Cerebellar tubers also occur and more commonly are seen in patients with a high cerebral tuber burden. The subependymal nodules often calcify. Another manifestation of tuberous sclerosis complex is subependymal giant cell astrocytomas, which commonly occur near the foramen of Monro. Figure 38-3 A 3-month-old with tuberous sclerosis complex and right occipital lobe intractable epilepsy. Overview and Imaging: Sturge-Weber syndrome, or encephalotrigeminal angiomatosis, is a phakomatosis characterized by a facial capillary vascular malformation or “port wine stain” in the territory of the trigeminal nerve, ipsilateral leptomeningeal angiomatosis, and angiomatosis of the choroid of the ipsilateral eye. Seizures usually are the initial neurological manifestation in the first year of life. Children with early onset of epilepsy are more likely to have hemiparesis, status epilepticus, and developmental delay compared with persons whose seizures start later in life.49 Imaging findings include leptomeningeal angiomatosis, enlargement of the choroid plexus, parenchymal atrophy, calvarial changes, and calcification (Fig. 38-4). Leptomeningeal angiomatosis and choroid plexus hyperplasia appear earlier in the course of the disease, whereas atrophy and calcification usually are evident later. Contrast-enhanced MRI is considered the most sensitive sequence for depicting leptomeningeal enhancement and revealing subtle bilateral hemispheric involvement, compensatory venous drainage, enlarged choroid plexus in the atria, and cerebellar and eye involvement.50–52 Figure 38-4 A 15-month-old boy with Sturge-Weber syndrome. Overview and Imaging: Mesial temporal sclerosis is encountered less frequently in children than in adults.53–56 The etiology of hippocampal sclerosis is unknown, but a link with febrile seizures has been suggested.57,58 Hippocampal sclerosis is characterized by neuronal loss and gliosis in the hippocampus.59 MRI features include atrophy and increased T2 and FLAIR signal in the hippocampus (Fig. 38-5).59–62 Other findings include loss of interdigitations of the hippocampal head,63 atrophy of the ipsilateral mamillary body and fornix,64 dilatation of the ipsilateral temporal horn, volume loss of the temporal lobe, and atrophy of the collateral white matter between the hippocampus and collateral sulcus.62 Functional imaging with FDG-PET usually reveals hypometabolic activity that is larger than the hippocampus abnormality65,66 and can identify mesial temporal metabolic abnormalities in patients with normal MRI.67 Epilepsy-associated tumors may account for up to two thirds of the surgical pathologic substrate72–75 in epilepsy surgery. These tumors originate in and develop from the cortex and present clinically with seizures. Tumors associated with epilepsy usually are associated with benign biological behavior and a low proliferation index. Only a small percentage of these tumors may undergo malignant transformation.76 The tumors include gangliogliomas, gangliocytomas, desmoplastic infantile gangliogliomas, dysembryoplastic neuroepithelial tumors (DNETs), pleomorphic xanthoastrocytomas (PXAs), and low-grade astrocytomas.77 FCD may be present concurrently. The most common clinical presentation of gangliogliomas is seizures, which often are complex, partial, and medically intractable.78 Tumors are larger in children than in adults,79 and they are located mostly in the temporomesial (50%) or temporolateral (29%) location.80 Gangliogliomas are composed of glia and neurons and may present as a solid mass in 43% of cases, a cyst in 5% of cases, and a mixed lesion in 52% of cases.76,81 Because they are cortical, they may cause remodeling of the adjacent bone. Most gangliogliomas (38%) are hypodense on computed tomography (CT), but isodense (15%), hyperdense (15%), or mixed density masses (32%) can be found,82 and calcification is seen in 30% to 50% of cases. On MRI, the solid part of the tumor may appear hypointense or isointense to gray matter on T1-weighted images and hyperintense on T2-weighted images (Fig. 38-6
Neuroimaging in Pediatric Epilepsy
Malformations of Cortical Development
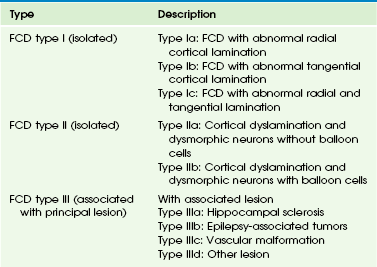
Focal Cortical Dysplasia
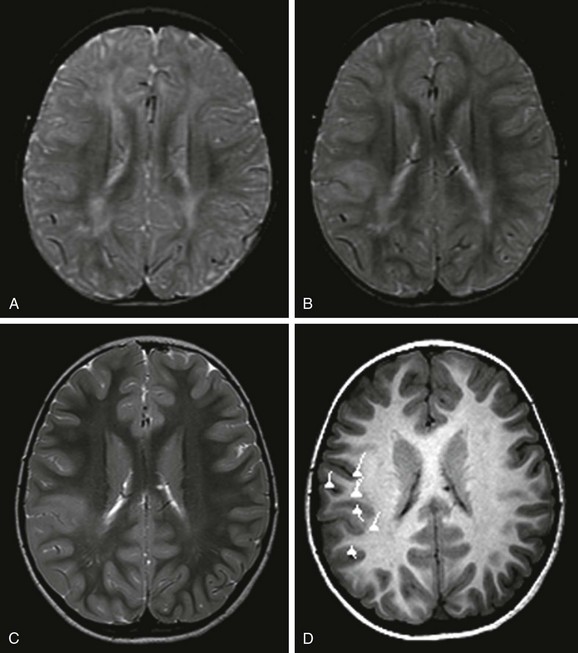
An axial T2-weighted image at 12 months (A) does not show a lesion. However, at 19 months (B) and 5 years of age (C), axial T2-weighted images demonstrate an abnormal signal in the cortex and subcortical white matter that is associated with blurring of the gray/white matter junction. D, Magnetoencephalography projected onto an axial T1-weighted image demonstrates that the dipoles, indicative of the epileptogenic zone, are located in and around the lesion. Surgical resection of the lesion and epileptogenic zone confirms the presence of focal cortical dysplasia type IIB.
Hemimegalencephaly
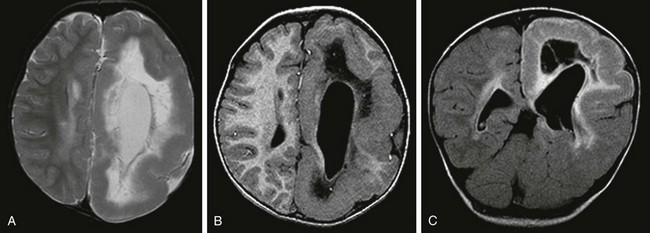
An axial T2-weighted image (A), an axial volumetric T1-weighted image (B), and a coronal fluid attenuated inversion recovery (FLAIR) image (C) demonstrate diffuse enlargement of the left cerebral hemisphere that is associated with an abnormally thick cortex, an abnormal sulcation and gyration pattern, and a dysmorphic and enlarged left lateral ventricle. An abnormal increased T2 and FLAIR signal and a reduced T1 signal in the white matter of the affected hemisphere also are noted.
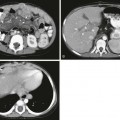
Tuberous Sclerosis Complex
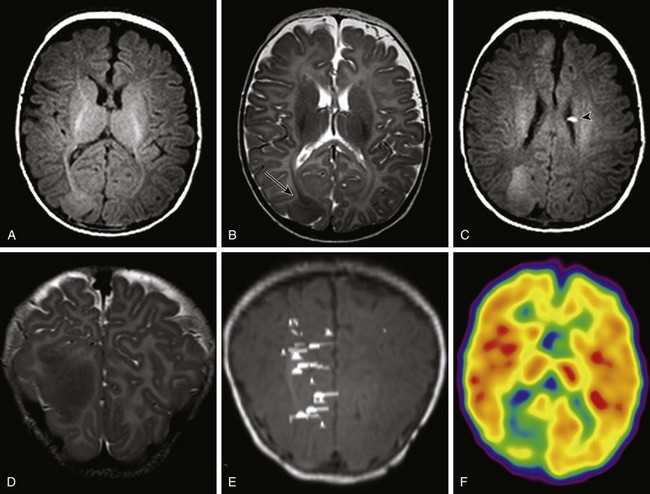
Axial T1-weighted images (A and C), an axial T2-weighted image (B), and a coronal T2-weighted image (D) indicate several cortical/subcortical tubers in the right frontal and occipital lobes (arrow) that demonstrate a high T1 and lower T2 signal. Several subependymal nodules also are present (arrowhead). E, Magnetoencephalography dipoles projected onto a coronal T1 image demonstrate a dipole cluster in the right occipital lobe. F, A fluorodeoxyglucose positron emission tomography study shows hypometabolism (blue and green) that is more extensive than the right occipital tuber.
Sturge-Weber Syndrome
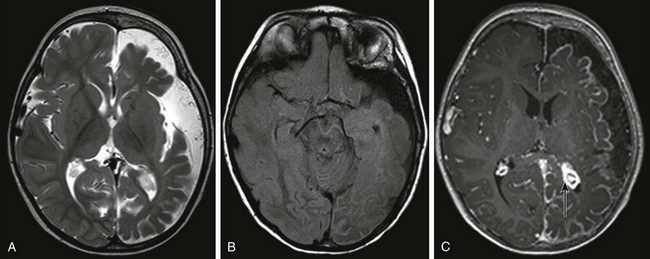
An axial T2-weighted image (A) and an axial fluid attenuated inversion recovery (FLAIR) image (B) show volume loss in the left cerebral hemisphere with enlargement of subarachnoid spaces and a high FLAIR signal in the left cerebral sulci. C, An axial T1-weighted postcontrast image shows pial enhancement over the left cerebral hemisphere, as well as an enlarged choroid plexus in the left trigone (arrow).
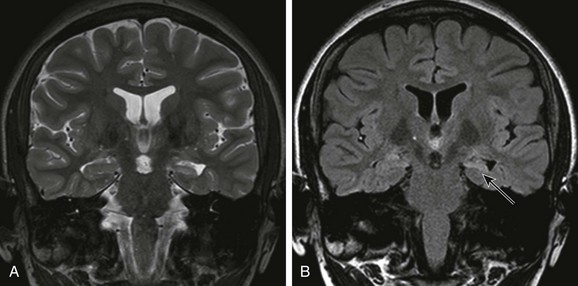
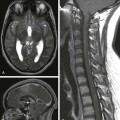
Mesial Temporal Sclerosis
Epilepsy-Associated Developmental Tumors
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Neuroimaging in Pediatric Epilepsy