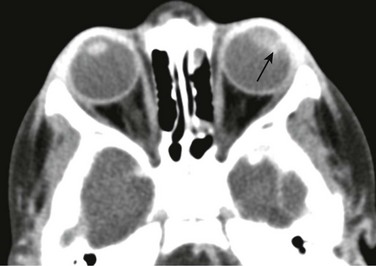
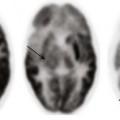
Chapter 7 Approximately two thirds of all retinoblastoma cases are unilateral, and one third are bilateral. Several classification systems have been developed for intraocular retinoblastoma, all of which are based on expected results of therapy and predicted salvage of the globe. The rapid evolution of newer retinoblastoma treatment has resulted in the replacement of the previously widely accepted Reese-Ellsworth classification, which is based on intraocular tumor staging and is used in tumor management after external beam radiation. A new International Classification of Retinoblastoma is based mainly on the natural history of retinoblastoma (early disease [group A] to late disease [group E]) and upon the extent of tumor seeding within the vitreous and subretinal space (Box 7-1). This classification is more applicable for patients treated with chemotherapy. Leukocoria in a young child that is confirmed by an ophthalmoscopic examination requires further evaluation. Imaging may reliably distinguish retinoblastoma from a host of other conditions that also may present with leukocoria (e.g., persistent hyperplastic primary vitreous, Toxascaris infection, Coats disease, and medulloepithelioma). In many retinoblastoma centers, eye ultrasonography has replaced orbital CT for initial disease assessment. Three-dimensional high-frequency ultrasound is sufficient for assessment of tumor and calcifications but is less suitable for the evaluation of extraocular spread. Retinoblastoma is readily visible on ultrasound as an echogenic irregular retinal mass with focal acoustic shadows. CT is an excellent tool for depicting ocular calcifications (Fig. 7-1, A), but its use in retinoblastoma evaluation has markedly decreased because of the associated radiation risks. MRI has become a relatively quick and convenient modality for the evaluation of retinoblastoma. A variety of tailored MR sequences are beneficial in illustrating the different features of these masses. Specifically, intraocular hemorrhages and calcifications are depicted best on gradient sequences, whereas the malignant, markedly cellular nature of these tumors (composed of immature retinoblasts) is confirmed on T2-weighted and DWI sequences (Fig. 7-1, B and C). Contrast-enhanced sequences provide important information that may affect prognosis and treatment options with respect to tumor spread into the optic nerve (Fig. 7-1, D), anterior chamber, or other adjacent orbital structures. In addition, MRI serves as a surveillance examination of the brain when monitoring for possible leptomeningeal spread of tumor or for the presence of retinoblastoma in more remote locations. Figure 7-1 Retinoblastoma of the orbits in three different patients. A medulloepithelioma is another pediatric malignant intraocular tumor that may present with leukocoria. This tumor is a rare embryonal type of neoplasm arising from the nonpigmented epithelial lining of the ciliary body. Patients usually are diagnosed in the first decade of life (at a mean age of 6 years); only rarely is medulloepithelioma seen in adults. Many imaging features of medulloepithelioma may closely resemble those of a retinoblastoma, such as presentation with a nodular enhancing intraocular mass, which occasionally will have calcifications (Fig. 7-2). A medulloepithelioma differs from a retinoblastoma mainly by its anterior location, but it may appear identical to a retinoblastoma when it is located in the vicinity of the optic nerve. Medulloepitheliomas have been divided into two types, teratoid and nonteratoid (diktyoma), based on their histologies. More complex teratoid medulloepitheliomas are composed of heteroplastic elements, including cartilage, which may have associated calcifications, whereas the nonteratoid diktyoma presents as a well-defined, noncalcified mass with associated diffuse contrast enhancement. Ocular manifestations of tuberous sclerosis include astrocytic hamartomas of the retina and optic disk. These lesions have a typical appearance on ophthalmologic examination; however, they may calcify as the patient ages and may in fact resemble drusen when located on the optic disk. On thin-section high-resolution T2-weighted imaging, hamartomas of tuberous sclerosis are visible as small hypointense nodules within the posterior globe (e-Fig. 7-3). e-Figure 7-3 Retinal hamartoma of tuberous sclerosis. Disc drusen are nonhamartomatous subretinal lesions without astrocytic hyperplasia that are associated with the presence of intrapapillary, partially calcified hyaline bodies that form concretions of unknown nature. Drusen likely are the most common etiology for congenital bilateral elevation of the optic nerve discs. Drusen may be detected on funduscopic evaluation or may be seen as an incidental finding on imaging. In both scenarios, it is important to establish the benign nature of disk elevation so as not to confuse drusen (which cause pseudopapilledema) with true papilledema. Equivocal results of ophthalmoscopic examination may lead to orbital imaging, which will either confirm the presence of disc drusen or detect the source of the increased intracranial pressure. Drusen may be diagnosed with use of ultrasound, appearing as foci of increased echogenicity, or drusen can be seen on noncontrast CT most often as bilateral punctuate calcifications within the optic nerve heads (e-Fig. 7-4). MRI demonstrates isolated mild protrusion of the optic discs into the vitreous without perioptic cerebrospinal fluid space enlargement or other imaging features of papilledema. Clinically, drusen are usually asymptomatic and only rarely may be associated with slowly progressive visual loss.
Orbital Neoplasia
Ocular Lesions
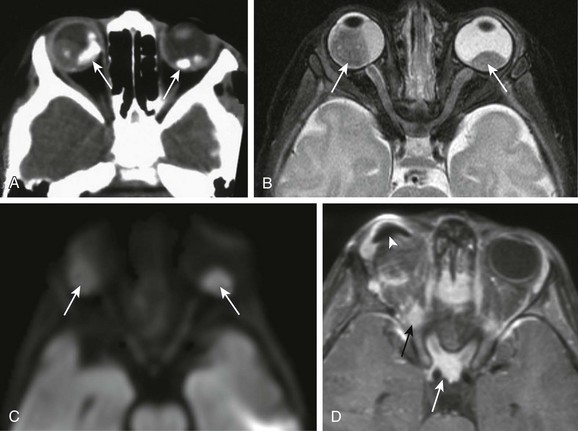
A, An orbital computed tomography scan demonstrates bilateral, markedly calcified orbital masses (arrows). Orbital magnetic resonance images (MRI), including an axial fat-saturated T2-weighted image (B) and a diffusion-weighted image (C), demonstrate bilateral, lobulated, T2-hypointense, retinal-based masses that exhibit diffusion restriction (arrows). D, In another patient with known recurrent retinoblastoma who has had right enucleation and prosthesis insertion (arrowhead), postcontrast T1-weighted fat-saturated MRI demonstrates fusiform enlargement and enhancement of the stump of the optic nerve sheath throughout its intraorbital (black arrow) and intracanalicular components, with associated posterior extension to the optic chiasm (white arrow).
Medulloepithelioma
Hereditary Orbital Hamartomatosis
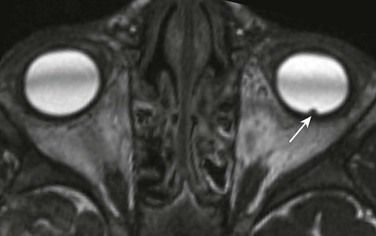
An axial constructive interference steady-state image of the orbit in a patient with known tuberous sclerosis shows small, punctate retinal hamartoma along the posterior wall of the left globe (arrow).
Drusen
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Orbital Neoplasia